Short Branch Attraction, the Fundamental Bipartition in Cellular Life, and Eukaryogenesis Amanda A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture Notes: the Mathematics of Phylogenetics
Lecture Notes: The Mathematics of Phylogenetics Elizabeth S. Allman, John A. Rhodes IAS/Park City Mathematics Institute June-July, 2005 University of Alaska Fairbanks Spring 2009, 2012, 2016 c 2005, Elizabeth S. Allman and John A. Rhodes ii Contents 1 Sequences and Molecular Evolution 3 1.1 DNA structure . .4 1.2 Mutations . .5 1.3 Aligned Orthologous Sequences . .7 2 Combinatorics of Trees I 9 2.1 Graphs and Trees . .9 2.2 Counting Binary Trees . 14 2.3 Metric Trees . 15 2.4 Ultrametric Trees and Molecular Clocks . 17 2.5 Rooting Trees with Outgroups . 18 2.6 Newick Notation . 19 2.7 Exercises . 20 3 Parsimony 25 3.1 The Parsimony Criterion . 25 3.2 The Fitch-Hartigan Algorithm . 28 3.3 Informative Characters . 33 3.4 Complexity . 35 3.5 Weighted Parsimony . 36 3.6 Recovering Minimal Extensions . 38 3.7 Further Issues . 39 3.8 Exercises . 40 4 Combinatorics of Trees II 45 4.1 Splits and Clades . 45 4.2 Refinements and Consensus Trees . 49 4.3 Quartets . 52 4.4 Supertrees . 53 4.5 Final Comments . 54 4.6 Exercises . 55 iii iv CONTENTS 5 Distance Methods 57 5.1 Dissimilarity Measures . 57 5.2 An Algorithmic Construction: UPGMA . 60 5.3 Unequal Branch Lengths . 62 5.4 The Four-point Condition . 66 5.5 The Neighbor Joining Algorithm . 70 5.6 Additional Comments . 72 5.7 Exercises . 73 6 Probabilistic Models of DNA Mutation 81 6.1 A first example . 81 6.2 Markov Models on Trees . 87 6.3 Jukes-Cantor and Kimura Models . -

Species Concepts Should Not Conflict with Evolutionary History, but Often Do
ARTICLE IN PRESS Stud. Hist. Phil. Biol. & Biomed. Sci. xxx (2008) xxx–xxx Contents lists available at ScienceDirect Stud. Hist. Phil. Biol. & Biomed. Sci. journal homepage: www.elsevier.com/locate/shpsc Species concepts should not conflict with evolutionary history, but often do Joel D. Velasco Department of Philosophy, University of Wisconsin-Madison, 5185 White Hall, 600 North Park St., Madison, WI 53719, USA Department of Philosophy, Building 90, Stanford University, Stanford, CA 94305, USA article info abstract Keywords: Many phylogenetic systematists have criticized the Biological Species Concept (BSC) because it distorts Biological Species Concept evolutionary history. While defences against this particular criticism have been attempted, I argue that Phylogenetic Species Concept these responses are unsuccessful. In addition, I argue that the source of this problem leads to previously Phylogenetic Trees unappreciated, and deeper, fatal objections. These objections to the BSC also straightforwardly apply to Taxonomy other species concepts that are not defined by genealogical history. What is missing from many previous discussions is the fact that the Tree of Life, which represents phylogenetic history, is independent of our choice of species concept. Some species concepts are consistent with species having unique positions on the Tree while others, including the BSC, are not. Since representing history is of primary importance in evolutionary biology, these problems lead to the conclusion that the BSC, along with many other species concepts, are unacceptable. If species are to be taxa used in phylogenetic inferences, we need a history- based species concept. Ó 2008 Elsevier Ltd. All rights reserved. When citing this paper, please use the full journal title Studies in History and Philosophy of Biological and Biomedical Sciences 1. -

Marsarchaeota Are an Aerobic Archaeal Lineage Abundant in Geothermal Iron Oxide Microbial Mats
Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats Authors: Zackary J. Jay, Jacob P. Beam, Mansur Dlakic, Douglas B. Rusch, Mark A. Kozubal, and William P. Inskeep This is a postprint of an article that originally appeared in Nature Microbiology on May 14, 2018. The final version can be found at https://dx.doi.org/10.1038/s41564-018-0163-1. Jay, Zackary J. , Jacob P. Beam, Mensur Dlakic, Douglas B. Rusch, Mark A. Kozubal, and William P. Inskeep. "Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats." Nature Microbiology 3, no. 6 (May 2018): 732-740. DOI: 10.1038/ s41564-018-0163-1. Made available through Montana State University’s ScholarWorks scholarworks.montana.edu Marsarchaeota are an aerobic archaeal lineage abundant in geothermal iron oxide microbial mats Zackary J. Jay1,4,7, Jacob P. Beam1,5,7, Mensur Dlakić2, Douglas B. Rusch3, Mark A. Kozubal1,6 and William P. Inskeep 1* The discovery of archaeal lineages is critical to our understanding of the universal tree of life and evolutionary history of the Earth. Geochemically diverse thermal environments in Yellowstone National Park provide unprecedented opportunities for studying archaea in habitats that may represent analogues of early Earth. Here, we report the discovery and character- ization of a phylum-level archaeal lineage proposed and herein referred to as the ‘Marsarchaeota’, after the red planet. The Marsarchaeota contains at least two major subgroups prevalent in acidic, microaerobic geothermal Fe(III) oxide microbial mats across a temperature range from ~50–80 °C. Metagenomics, single-cell sequencing, enrichment culturing and in situ transcrip- tional analyses reveal their biogeochemical role as facultative aerobic chemoorganotrophs that may also mediate the reduction of Fe(III). -

Phylogenetic Comparative Methods: a User's Guide for Paleontologists
Phylogenetic Comparative Methods: A User’s Guide for Paleontologists Laura C. Soul - Department of Paleobiology, National Museum of Natural History, Smithsonian Institution, Washington, DC, USA David F. Wright - Division of Paleontology, American Museum of Natural History, Central Park West at 79th Street, New York, New York 10024, USA and Department of Paleobiology, National Museum of Natural History, Smithsonian Institution, Washington, DC, USA Abstract. Recent advances in statistical approaches called Phylogenetic Comparative Methods (PCMs) have provided paleontologists with a powerful set of analytical tools for investigating evolutionary tempo and mode in fossil lineages. However, attempts to integrate PCMs with fossil data often present workers with practical challenges or unfamiliar literature. In this paper, we present guides to the theory behind, and application of, PCMs with fossil taxa. Based on an empirical dataset of Paleozoic crinoids, we present example analyses to illustrate common applications of PCMs to fossil data, including investigating patterns of correlated trait evolution, and macroevolutionary models of morphological change. We emphasize the importance of accounting for sources of uncertainty, and discuss how to evaluate model fit and adequacy. Finally, we discuss several promising methods for modelling heterogenous evolutionary dynamics with fossil phylogenies. Integrating phylogeny-based approaches with the fossil record provides a rigorous, quantitative perspective to understanding key patterns in the history of life. 1. Introduction A fundamental prediction of biological evolution is that a species will most commonly share many characteristics with lineages from which it has recently diverged, and fewer characteristics with lineages from which it diverged further in the past. This principle, which results from descent with modification, is one of the most basic in biology (Darwin 1859). -

Lineages, Splits and Divergence Challenge Whether the Terms Anagenesis and Cladogenesis Are Necessary
Biological Journal of the Linnean Society, 2015, , – . With 2 figures. Lineages, splits and divergence challenge whether the terms anagenesis and cladogenesis are necessary FELIX VAUX*, STEVEN A. TREWICK and MARY MORGAN-RICHARDS Ecology Group, Institute of Agriculture and Environment, Massey University, Palmerston North, New Zealand Received 3 June 2015; revised 22 July 2015; accepted for publication 22 July 2015 Using the framework of evolutionary lineages to separate the process of evolution and classification of species, we observe that ‘anagenesis’ and ‘cladogenesis’ are unnecessary terms. The terms have changed significantly in meaning over time, and current usage is inconsistent and vague across many different disciplines. The most popular definition of cladogenesis is the splitting of evolutionary lineages (cessation of gene flow), whereas anagenesis is evolutionary change between splits. Cladogenesis (and lineage-splitting) is also regularly made synonymous with speciation. This definition is misleading as lineage-splitting is prolific during evolution and because palaeontological studies provide no direct estimate of gene flow. The terms also fail to incorporate speciation without being arbitrary or relative, and the focus upon lineage-splitting ignores the importance of divergence, hybridization, extinction and informative value (i.e. what is helpful to describe as a taxon) for species classification. We conclude and demonstrate that evolution and species diversity can be considered with greater clarity using simpler, more transparent terms than anagenesis and cladogenesis. Describing evolution and taxonomic classification can be straightforward, and there is no need to ‘make words mean so many different things’. © 2015 The Linnean Society of London, Biological Journal of the Linnean Society, 2015, 00, 000–000. -

The Phagotrophic Origin of Eukaryotes and Phylogenetic Classification Of
International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 DOI: 10.1099/ijs.0.02058-0 The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa Department of Zoology, T. Cavalier-Smith University of Oxford, South Parks Road, Oxford OX1 3PS, UK Tel: j44 1865 281065. Fax: j44 1865 281310. e-mail: tom.cavalier-smith!zoo.ox.ac.uk Eukaryotes and archaebacteria form the clade neomura and are sisters, as shown decisively by genes fragmented only in archaebacteria and by many sequence trees. This sisterhood refutes all theories that eukaryotes originated by merging an archaebacterium and an α-proteobacterium, which also fail to account for numerous features shared specifically by eukaryotes and actinobacteria. I revise the phagotrophy theory of eukaryote origins by arguing that the essentially autogenous origins of most eukaryotic cell properties (phagotrophy, endomembrane system including peroxisomes, cytoskeleton, nucleus, mitosis and sex) partially overlapped and were synergistic with the symbiogenetic origin of mitochondria from an α-proteobacterium. These radical innovations occurred in a derivative of the neomuran common ancestor, which itself had evolved immediately prior to the divergence of eukaryotes and archaebacteria by drastic alterations to its eubacterial ancestor, an actinobacterial posibacterium able to make sterols, by replacing murein peptidoglycan by N-linked glycoproteins and a multitude of other shared neomuran novelties. The conversion of the rigid neomuran wall into a flexible surface coat and the associated origin of phagotrophy were instrumental in the evolution of the endomembrane system, cytoskeleton, nuclear organization and division and sexual life-cycles. Cilia evolved not by symbiogenesis but by autogenous specialization of the cytoskeleton. -

Phylogeny Codon Models • Last Lecture: Poor Man’S Way of Calculating Dn/Ds (Ka/Ks) • Tabulate Synonymous/Non-Synonymous Substitutions • Normalize by the Possibilities
Phylogeny Codon models • Last lecture: poor man’s way of calculating dN/dS (Ka/Ks) • Tabulate synonymous/non-synonymous substitutions • Normalize by the possibilities • Transform to genetic distance KJC or Kk2p • In reality we use codon model • Amino acid substitution rates meet nucleotide models • Codon(nucleotide triplet) Codon model parameterization Stop codons are not allowed, reducing the matrix from 64x64 to 61x61 The entire codon matrix can be parameterized using: κ kappa, the transition/transversionratio ω omega, the dN/dS ratio – optimizing this parameter gives the an estimate of selection force πj the equilibrium codon frequency of codon j (Goldman and Yang. MBE 1994) Empirical codon substitution matrix Observations: Instantaneous rates of double nucleotide changes seem to be non-zero There should be a mechanism for mutating 2 adjacent nucleotides at once! (Kosiol and Goldman) • • Phylogeny • • Last lecture: Inferring distance from Phylogenetic trees given an alignment How to infer trees and distance distance How do we infer trees given an alignment • • Branch length Topology d 6-p E 6'B o F P Edo 3 vvi"oH!.- !fi*+nYolF r66HiH- .) Od-:oXP m a^--'*A ]9; E F: i ts X o Q I E itl Fl xo_-+,<Po r! UoaQrj*l.AP-^PA NJ o - +p-5 H .lXei:i'tH 'i,x+<ox;+x"'o 4 + = '" I = 9o FF^' ^X i! .poxHo dF*x€;. lqEgrE x< f <QrDGYa u5l =.ID * c 3 < 6+6_ y+ltl+5<->-^Hry ni F.O+O* E 3E E-f e= FaFO;o E rH y hl o < H ! E Y P /-)^\-B 91 X-6p-a' 6J. -

Diagnosis and Differentiation of the Order Primates
YEARBOOK OF PHYSICAL ANTHROPOLOGY 30:75-105 (1987) Diagnosis and Differentiation of the Order Primates FREDERICK S. SZALAY, ALFRED L. ROSENBERGER, AND MARIAN DAGOSTO Department of Anthropolog* Hunter College, City University of New York, New York, New York 10021 (F.S.S.); University of Illinois, Urbanq Illinois 61801 (A.L. R.1; School of Medicine, Johns Hopkins University/ Baltimore, h4D 21218 (M.B.) KEY WORDS Semiorders Paromomyiformes and Euprimates, Suborders Strepsirhini and Haplorhini, Semisuborder Anthropoidea, Cranioskeletal morphology, Adapidae, Omomyidae, Grades vs. monophyletic (paraphyletic or holophyletic) taxa ABSTRACT We contrast our approach to a phylogenetic diagnosis of the order Primates, and its various supraspecific taxa, with definitional proce- dures. The order, which we divide into the semiorders Paromomyiformes and Euprimates, is clearly diagnosable on the basis of well-corroborated informa- tion from the fossil record. Lists of derived features which we hypothesize to have been fixed in the first representative species of the Primates, Eupri- mates, Strepsirhini, Haplorhini, and Anthropoidea, are presented. Our clas- sification of the order includes both holophyletic and paraphyletic groups, depending on the nature of the available evidence. We discuss in detail the problematic evidence of the basicranium in Paleo- gene primates and present new evidence for the resolution of previously controversial interpretations. We renew and expand our emphasis on postcra- nial analysis of fossil and living primates to show the importance of under- standing their evolutionary morphology and subsequent to this their use for understanding taxon phylogeny. We reject the much advocated %ladograms first, phylogeny next, and scenario third” approach which maintains that biologically founded character analysis, i.e., functional-adaptive analysis and paleontology, is irrelevant to genealogy hypotheses. -
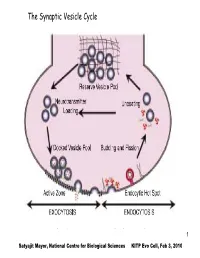
The Synaptic Vesicle Cycle
The Synaptic Vesicle Cycle 1 Satyajit Mayor, National Centre for Biological Sciences KITP Evo Cell, Feb 3, 2010 The Eukaryotic Commandments: T. Cavalier-Smith (i)origin of the endomembrane system (ER, Golgi and lysosomes) and coated- vesicle budding and fusion, including endocytosis and exocytosis; (ii) origin of the cytoskeleton, centrioles, cilia and associated molecular motors; (iii) origin of the nucleus, nuclear pore complex and trans-envelope protein and RNA transport; (iv) origin of linear chromosomes with plural replicons, centromeres and telomeres; (v) origin of novel cell-cycle controls and mitotic segregation; (vi) origin of sex (syngamy, nuclear fusion and meiosis); (vii) origin of peroxisomes; (ix) origin of mitochondria; (viii) novel patterns of rRNAprocessing using small nucleolar-ribonucleoproteins (snoRNPs); (x) origin of spliceosomal introns. 2 EM of Chemical synapse mitochondria Active Zone Figure 5.3, Bear, 2001 3 T. Cavalier-Smith International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 Fig. 1. The bacterial origins of eukaryotes as a two-stage process. I phase: The ancestors of eukaryotes, the stem neomura, are shared with archaebacteria and evolved during the neomuran revolution, in which N-linked glycoproteins replaced murein peptidoglycan and 18 other suites of characters changed radically through adaptation of an ancestral actinobacterium to thermophily, as discussed in detail by Cavalier- Smith (2002a). 4 T. Cavalier-Smith International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 Fig. 1. The bacterial origins of eukaryotes as a two-stage process. II phase: In the next phase, archaebacteria and eukaryotes diverged dramatically. Archaebacteria retained the wall and therefore their general bacterial cell and genetic organization, but became adapted to even hotter and more acidic environments by substituting prenyl ether lipids for the ancestral acyl esters and making new acid- resistant flagellar shafts (Cavalier-Smith, 2002a). -

A Revised Taxonomy of the Iguanodont Dinosaur Genera and Species
ARTICLE IN PRESS + MODEL Cretaceous Research xx (2007) 1e25 www.elsevier.com/locate/CretRes A revised taxonomy of the iguanodont dinosaur genera and species Gregory S. Paul 3109 North Calvert Station, Side Apartment, Baltimore, MD 21218-3807, USA Received 20 April 2006; accepted in revised form 27 April 2007 Abstract Criteria for designating dinosaur genera are inconsistent; some very similar species are highly split at the generic level, other anatomically disparate species are united at the same rank. Since the mid-1800s the classic genus Iguanodon has become a taxonomic grab-bag containing species spanning most of the Early Cretaceous of the northern hemisphere. Recently the genus was radically redesignated when the type was shifted from nondiagnostic English Valanginian teeth to a complete skull and skeleton of the heavily built, semi-quadrupedal I. bernissartensis from much younger Belgian sediments, even though the latter is very different in form from the gracile skeletal remains described by Mantell. Currently, iguanodont remains from Europe are usually assigned to either robust I. bernissartensis or gracile I. atherfieldensis, regardless of lo- cation or stage. A stratigraphic analysis is combined with a character census that shows the European iguanodonts are markedly more morpho- logically divergent than other dinosaur genera, and some appear phylogenetically more derived than others. Two new genera and a new species have been or are named for the gracile iguanodonts of the Wealden Supergroup; strongly bipedal Mantellisaurus atherfieldensis Paul (2006. Turning the old into the new: a separate genus for the gracile iguanodont from the Wealden of England. In: Carpenter, K. (Ed.), Horns and Beaks: Ceratopsian and Ornithopod Dinosaurs. -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

Distribution of Arsenite-Oxidizing Bacteria and Its Correlation with Environmental Factors in Geothermal Areas of Tengchong, Yunnan, China
E3S Web of Conferences 98, 02002 (2019) https://doi.org/10.1051/e3sconf/20199802002 WRI-16 Distribution of arsenite-oxidizing bacteria and its correlation with environmental factors in geothermal areas of Tengchong, Yunnan, China Ping Li1,*, Dawei Jiang1, and Zhou Jiang1,2 1State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan, China 2School of Environmental Studies, China University of Geosciences, Wuhan, China Abstract. Arsenic (As) is an ubiquitous constituent in geothermal water. Arsenite (AsIII) is oxidized via microbial processes as the waters equilibrate with oxygen in the geothermal effluent. The distribution of arsenite oxdizing bacteria and its correlation with environment factors were studied in Tengchong geothermal areas of Yunnan, China. A total of 230 aioA clone sequences were obtained and these sequences were affiliated with four phyla: Betaproteobacteria, Alphaproteobacteria, Deinococcus- Thermus and Aquificae. Temperature was negatively correlated with aioA diversity and was the only environment factor that had correlation with diversity index. Betaproteobacteria was mainly distributed in low temperature (T = 28 to 43 oC) and circumneutral or light alkaline (pH = 7 to 9) springs; Alphaproteobacteria was mainly predominant in low pH (pH = 3.3 to 3.6) springs; Deinococcus-Thermus and Aquificae mainly inhabited in high temperature (T=55 to 78 oC) springs with a wide range of pH. Usually, Deinococcus-Thermus was dominant when springs had a pH within 4.0 to 8.0. Aquificae was dominated in springs with pH > 8.0 or pH < 4.0. 1 Diversity of aioA gene A total of 230 aioA gene clone sequences from 10 sample sites were subjected to sequence similarity analysis.