Supplementary Figure 1. Sleep Traits Are Phenotypically and Genetically Correlated in Men and Women
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Itcs) in the Mouse Amygdala of Tshz1 Mutants Correlates with Fear, Depression, and Social Interaction Phenotypes
1160 • The Journal of Neuroscience, January 31, 2018 • 38(5):1160–1177 Development/Plasticity/Repair Loss of Intercalated Cells (ITCs) in the Mouse Amygdala of Tshz1 Mutants Correlates with Fear, Depression, and Social Interaction Phenotypes X Jeffrey Kuerbitz,1 Melinda Arnett,5 Sarah Ehrman,1 XMichael T. Williams,3 XCharles V. Vorhees,3 X Simon E. Fisher,6,7 Alistair N. Garratt,8 XLouis J. Muglia,5 Ronald R. Waclaw,1,4 and XKenneth Campbell1,2 Divisions of 1Developmental Biology, 2Neurosurgery, 3Neurology, 4Experimental Hematology and Cancer Biology, 5Center for Prevention of Preterm Birth, Perinatal Institute, Cincinnati Children’s Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, OH 45229, 6Language and Genetics Department, Max Planck Institute for Psycholinguistics, 6500 AH Nijmegen, The Netherlands, 7Donders Institute for Brain, Cognition and Behaviour, Radboud University, Nijmegen, The Netherlands, and 8Institute of Cell Biology and Neurobiology, Center for Anatomy, Charite´ University Hospital Berlin, 10117 Berlin, Germany The intercalated cells (ITCs) of the amygdala have been shown to be critical regulatory components of amygdalar circuits, which control appropriate fear responses. Despite this, the molecular processes guiding ITC development remain poorly understood. Here we establish the zinc finger transcription factor Tshz1 as a marker of ITCs during their migration from the dorsal lateral ganglionic eminence through maturity. Using germline and conditional knock-out (cKO) mouse models, we show that Tshz1 is required for the proper migration and differentiation of ITCs. In the absence of Tshz1, migrating ITC precursors fail to settle in their stereotypical locations encapsulating the lateral amygdala and BLA. Furthermore, they display reductions in the ITC marker Foxp2 and ectopic persistence of the dorsal lateral ganglionic eminence marker Sp8. -

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -

Open Dogan Phdthesis Final.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science ELUCIDATING BIOLOGICAL FUNCTION OF GENOMIC DNA WITH ROBUST SIGNALS OF BIOCHEMICAL ACTIVITY: INTEGRATIVE GENOME-WIDE STUDIES OF ENHANCERS A Dissertation in Biochemistry, Microbiology and Molecular Biology by Nergiz Dogan © 2014 Nergiz Dogan Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2014 ii The dissertation of Nergiz Dogan was reviewed and approved* by the following: Ross C. Hardison T. Ming Chu Professor of Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee David S. Gilmour Professor of Molecular and Cell Biology Anton Nekrutenko Professor of Biochemistry and Molecular Biology Robert F. Paulson Professor of Veterinary and Biomedical Sciences Philip Reno Assistant Professor of Antropology Scott B. Selleck Professor and Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School iii ABSTRACT Genome-wide measurements of epigenetic features such as histone modifications, occupancy by transcription factors and coactivators provide the opportunity to understand more globally how genes are regulated. While much effort is being put into integrating the marks from various combinations of features, the contribution of each feature to accuracy of enhancer prediction is not known. We began with predictions of 4,915 candidate erythroid enhancers based on genomic occupancy by TAL1, a key hematopoietic transcription factor that is strongly associated with gene induction in erythroid cells. Seventy of these DNA segments occupied by TAL1 (TAL1 OSs) were tested by transient transfections of cultured hematopoietic cells, and 56% of these were active as enhancers. Sixty-six TAL1 OSs were evaluated in transgenic mouse embryos, and 65% of these were active enhancers in various tissues. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

An Unbiased Reconstruction of the T Helper Cell Type 2 Differentiation Network
bioRxiv preprint doi: https://doi.org/10.1101/196022; this version posted October 4, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. An unbiased reconstruction of the T helper cell type 2 differentiation network 1,3 1 1 1 1 Authors: Johan Henriksson , Xi Chen , Tomás Gomes , Kerstin Meyer , Ricardo Miragaia , 4 1 4 1 1,2,* Ubaid Ullah , Jhuma Pramanik , Riita Lahesmaa , Kosuke Yusa , Sarah A Teichmann Affiliations: 1 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SA, United Kingdom 2 EMBL-European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, United Kingdom 3 Karolinska Institutet, Department. of Biosciences and Nutrition, Hälsovägen 7, Novum, SE-141 83, Huddinge, Sweden 4 Turku Centre for Biotechnology, Tykistokatu 6 FI-20520, Turku, Finland *To whom correspondence should be addressed: [email protected] Tomas: [email protected] Ricardo: [email protected] Ubaid Ullah: [email protected] Jhuma: [email protected] -

FOXF2 Differentially Regulates Expression of Metabolic Genes In
Trends in Diabetes and Metabolism Research Article ISSN: 2631-9926 FOXF2 differentially regulates expression of metabolic genes in non-cancerous and cancerous breast epithelial cells Pang-Kuo Lo* Department of Biochemistry and Molecular Biology, Greenebaum Cancer Center, University of Maryland School of Medicine, USA Abstract Forkhead box F2 (FOXF2) functions as a transcription factor and is critically involved in programming organogenesis and regulating epithelial-to-mesenchymal transition (EMT) and cell proliferation. We recently have revealed that FOXF2 can exert distinct functional effects on different molecular subtypes of breast cancer. We found that FOXF2 expression is epigenetically silenced in luminal breast cancers due to its tumor-suppressive role in DNA replication regulation. In contrast, FOXF2 is overexpressed in basal-like triple-negative breast cancers (TNBCs) due to its oncogenic role in promoting EMT. Although our and other studies have shown that FOXF2 dysregulation is critical for tumorigenesis of various tissue types, the role of FOXF2 in metabolic rewiring of cancer remains unknown. In this study, we analyzed our previous microarray data to understand the metabolic role of FOXF2 in non-cancerous and cancerous breast epithelial cells. Our studies showed that in non-cancerous breast epithelial cells FOXF2 can also play a dual role either in tumor suppression or in tumor promotion through regulating expression of tumor-suppressive and oncogenic metabolic genes. Furthermore, we found that FOXF2-regulated metabolic genes are not conserved between non-cancerous and cancerous breast epithelial cells and FOXF2 is involved in metabolic rewiring in breast cancer cells. This is the first report to explore the metabolic function of FOXF2 in breast cancer. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Did Adam and Eve Have the Chromosome 2 Fusion?
Stankiewicz Molecular Cytogenetics (2016) 9:72 DOI 10.1186/s13039-016-0283-3 HYPOTHESIS Open Access One pedigree we all may have come from – did Adam and Eve have the chromosome 2fusion? Paweł Stankiewicz Abstract Background: In contrast to Great Apes, who have 48 chromosomes, modern humans and likely Neandertals and Denisovans have and had, respectively, 46 chromosomes. The reduction in chromosome number was caused by the head-to-head fusion of two ancestral chromosomes to form human chromosome 2 (HSA2) and may have contributed to the reproductive barrier with Great Apes. Results: Next generation sequencing and molecular clock analyses estimated that this fusion arose prior to our last common ancestor with Neandertal and Denisovan hominins ~ 0.74 - 4.5 million years ago. Hypotheses: I propose that, unlike recurrent Robertsonian translocations in humans, the HSA2 fusion was a single nonrecurrent event that spread through a small polygamous clan population bottleneck. Its heterozygous to homozygous conversion, fixation, and accumulation in the succeeding populations was likely facilitated by an evolutionary advantage through the genomic loss rather than deregulation of expression of the gene(s) flanking the HSA2 fusion site at 2q13. Conclusions: The origin of HSA2 might have been a critical evolutionary event influencing higher cognitive functions in various early subspecies of hominins. Next generation sequencing of Homo heidelbergensis and Homo erectus genomes and complete reconstruction of DNA sequence of the orthologous subtelomeric -
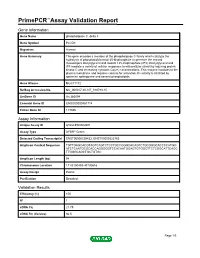
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name phospholipase C, delta 3 Gene Symbol PLCD3 Organism Human Gene Summary This gene encodes a member of the phospholipase C family which catalyze the hydrolysis of phosphatidylinositol 45-bisphosphate to generate the second messengers diacylglycerol and inositol 145-trisphosphate (IP3). Diacylglycerol and IP3 mediate a variety of cellular responses to extracellular stimuli by inducing protein kinase C and increasing cytosolic Ca(2+) concentrations. This enzyme localizes to the plasma membrane and requires calcium for activation. Its activity is inhibited by spermine sphingosine and several phospholipids. Gene Aliases MGC71172 RefSeq Accession No. NC_000017.10, NT_010783.15 UniGene ID Hs.380094 Ensembl Gene ID ENSG00000161714 Entrez Gene ID 113026 Assay Information Unique Assay ID qHsaCED0002601 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000539433, ENST00000322765 Amplicon Context Sequence TGTTGAGCACGTAGTCAGTCTCCTGCCGGGCACAGTCTGCGGGCACCCCATGG ATCTCAATGCGCACCAGGGGGTCCACAATGGAGTGTGGCTTCTCGGCATTCAGC TTGGGCAGCTGCTGTGC Amplicon Length (bp) 94 Chromosome Location 17:43190493-43190616 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 1 cDNA Cq 21.75 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 23.62 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLCD3, Human Amplification Plot Amplification of cDNA generated from -

Neurological Disorders, Genetic Correlations, and the Role of Exome Sequencing
Journal of Translational Science Review Article ISSN: 2059-268X Neurological disorders, genetic correlations, and the role of exome sequencing Tony L Brown1* and Theresa M Meloche2 1Columbia University, USA 2Advanced Research and Human Development Institute, USA Abstract Genomic information access and utilization by researchers and clinicians have barely begun the journey for fulfillment of their full potential in the research and clinical arenas. Exciting is the potential depth and breadth of research, clinical applications, and more personalized medicine, that remain on the horizon. Exome sequencing has clarified the responsibilities of over 130 genes, greatly expanding the medical genetics database and enabling the development of orphan disease- based pharmaceuticals. The focus of our research efforts was to review several literature sources related to rare genomic disease research and exome sequencing, as well as to review the new research and diagnostic strategies that were utilized. Using a systems approach, under discussion are neurology, neuropathy, and the central nervous and musculoskeletal systems. Also discussed will be the topics of inborn errors of metabolism, and the genetic targets related to developmental delay. Recommendations for future research will also be discussed. Exome sequencing neuronal ceroid lipofuscinoses the most common group of inherited neurological degenerative disorders [6]. Whether examining the A review of new strategies for rare genomic disease research mitochondrial defect implicated in prenatal ventriculomegaly