1 Title: Genomic and Transcriptomic Characterisation of Undifferentiated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptome Analyses of Rhesus Monkey Pre-Implantation Embryos Reveal A
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Transcriptome analyses of rhesus monkey pre-implantation embryos reveal a reduced capacity for DNA double strand break (DSB) repair in primate oocytes and early embryos Xinyi Wang 1,3,4,5*, Denghui Liu 2,4*, Dajian He 1,3,4,5, Shengbao Suo 2,4, Xian Xia 2,4, Xiechao He1,3,6, Jing-Dong J. Han2#, Ping Zheng1,3,6# Running title: reduced DNA DSB repair in monkey early embryos Affiliations: 1 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 2 Key Laboratory of Computational Biology, CAS Center for Excellence in Molecular Cell Science, Collaborative Innovation Center for Genetics and Developmental Biology, Chinese Academy of Sciences-Max Planck Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China 3 Yunnan Key Laboratory of Animal Reproduction, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China 4 University of Chinese Academy of Sciences, Beijing, China 5 Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650204, China 6 Primate Research Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China * Xinyi Wang and Denghui Liu contributed equally to this work 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press # Correspondence: Jing-Dong J. Han, Email: [email protected]; Ping Zheng, Email: [email protected] Key words: rhesus monkey, pre-implantation embryo, DNA damage 2 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Pre-implantation embryogenesis encompasses several critical events including genome reprogramming, zygotic genome activation (ZGA) and cell fate commitment. -

Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
908 J Med Genet: first published as 10.1136/jmg.2004.023184 on 9 December 2004. Downloaded from ORIGINAL ARTICLE Detection of aneuploidies by paralogous sequence quantification S Deutsch, U Choudhury, G Merla, C Howald, A Sylvan, S E Antonarakis ............................................................................................................................... J Med Genet 2004;41:908–915. doi: 10.1136/jmg.2004.023184 Background: Chromosomal aneuploidies are a common cause of congenital disorders associated with cognitive impairment and multiple dysmorphic features. Pre-natal diagnosis of aneuploidies is most See end of article for commonly performed by the karyotyping of fetal cells obtained by amniocentesis or chorionic villus authors’ affiliations sampling, but this method is labour intensive and requires about 14 days to complete. ....................... Methods: We have developed a PCR based method for the detection of targeted chromosome number Correspondence to: abnormalities termed paralogous sequence quantification (PSQ), based on the use of paralogous genes. Professor Stylianos E Paralogous sequences have a high degree of sequence identity, but accumulate nucleotide substitutions in Antonarakis, Department a locus specific manner. These sequence differences, which we term paralogous sequence mismatches of Genetic Medicine and Development, University of (PSMs), can be quantified using pyrosequencing technology, to estimate the relative dosage between Geneva Medical School, different chromosomes. We designed 10 assays for the detection of trisomies of chromosomes 13, 18, and GE 1211, Geneva, 21 and sex chromosome aneuploidies. Switzerland; Stylianos. antonarakis@medecine. Results: We evaluated the performance of this method on 175 DNAs, highly enriched for abnormal unige.ch samples. A correct and unambiguous diagnosis was given for 119 out of 120 aneuploid samples as well as for all the controls. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -
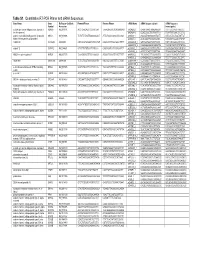
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

STK24 Antibody - C-Terminal Region Rabbit Polyclonal Antibody Catalog # AI16132
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 STK24 Antibody - C-terminal region Rabbit Polyclonal Antibody Catalog # AI16132 Specification STK24 Antibody - C-terminal region - Product Information Application WB Primary Accession Q9Y6E0 Other Accession NP_003567 Reactivity Human Host Rabbit Clonality Polyclonal Calculated MW 48kDa KDa STK24 Antibody - C-terminal region - Additional Information Host: Rabbit Target Name: STK24 Gene ID 8428 Sample Tissue: 293T Whole Cell lysates Antibody Dilution: 1.0μg/ml Alias Symbol STK24, MST3, STK3, Other Names STK24 Antibody - C-terminal region - Serine/threonine-protein kinase 24, Background 2.7.11.1, Mammalian STE20-like protein kinase 3, MST-3, STE20-like kinase MST3, Serine/threonine-protein kinase that acts on Serine/threonine-protein kinase 24 36 kDa both serine and threonine residues and subunit, Mammalian STE20-like protein promotes apoptosis in response to stress kinase 3 N-terminal, MST3/N, stimuli and caspase activation. Mediates Serine/threonine-protein kinase 24 12 kDa oxidative-stress- induced cell death by subunit, Mammalian STE20-like protein modulating phosphorylation of JNK1-JNK2 kinase 3 C-terminal, MST3/C, STK24, MST3, (MAPK8 and MAPK9), p38 (MAPK11, MAPK12, STK3 MAPK13 and MAPK14) during oxidative stress. Plays a role in a staurosporine-induced Format caspase- independent apoptotic pathway by Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and regulating the nuclear translocation of AIFM1 2% sucrose. and ENDOG and the DNase activity associated with ENDOG. Phosphorylates STK38L on Reconstitution & Storage 'Thr-442' and stimulates its kinase activity. Add 50 &mu, l of distilled water. -

MST3 (STK24) (NM 001032296) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC209117L4V MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Symbol: STK24 Synonyms: HEL-S-95; MST3; MST3B; STE20; STK3 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_001032296 ORF Size: 1293 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC209117). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_001032296.2 RefSeq Size: 4624 bp RefSeq ORF: 1296 bp Locus ID: 8428 UniProt ID: Q9Y6E0, Q5U0E6, Q6P0Y1 Protein Families: Druggable Genome, Protein Kinase MW: 47.7 kDa Gene Summary: This gene encodes a serine/threonine protein kinase that functions upstream of mitogen- activated protein kinase (MAPK) signaling. The encoded protein is cleaved into two chains by caspases; the N-terminal fragment (MST3/N) translocates to the nucleus and promotes programmed cells death. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

STRIPAK Complexes in Cell Signaling and Cancer
Oncogene (2016), 1–9 © 2016 Macmillan Publishers Limited All rights reserved 0950-9232/16 www.nature.com/onc REVIEW STRIPAK complexes in cell signaling and cancer Z Shi1,2, S Jiao1 and Z Zhou1,3 Striatin-interacting phosphatase and kinase (STRIPAK) complexes are striatin-centered multicomponent supramolecular structures containing both kinases and phosphatases. STRIPAK complexes are evolutionarily conserved and have critical roles in protein (de) phosphorylation. Recent studies indicate that STRIPAK complexes are emerging mediators and regulators of multiple vital signaling pathways including Hippo, MAPK (mitogen-activated protein kinase), nuclear receptor and cytoskeleton remodeling. Different types of STRIPAK complexes are extensively involved in a variety of fundamental biological processes ranging from cell growth, differentiation, proliferation and apoptosis to metabolism, immune regulation and tumorigenesis. Growing evidence correlates dysregulation of STRIPAK complexes with human diseases including cancer. In this review, we summarize the current understanding of the assembly and functions of STRIPAK complexes, with a special focus on cell signaling and cancer. Oncogene advance online publication, 15 February 2016; doi:10.1038/onc.2016.9 INTRODUCTION in the central nervous system and STRN4 is mostly abundant in Recent proteomic studies identified a group of novel multi- the brain and lung, whereas STRN3 is ubiquitously expressed in 5–9 component complexes named striatin (STRN)-interacting phos- almost all tissues. STRNs share a -

Mouse Farp1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Farp1 Knockout Project (CRISPR/Cas9) Objective: To create a Farp1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Farp1 gene (NCBI Reference Sequence: NM_134082 ; Ensembl: ENSMUSG00000025555 ) is located on Mouse chromosome 14. 27 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 27 (Transcript: ENSMUST00000026635). Exon 2 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from the coding region. Exon 2 covers 5.44% of the coding region. The size of effective KO region: ~194 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 27 Legends Exon of mouse Farp1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 2 is aligned with itself to determine if there are tandem repeats. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

STRIPAK Directs PP2A Activity Toward MAP4K4 to Promote Oncogenic Transformation
bioRxiv preprint doi: https://doi.org/10.1101/823096; this version posted October 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation Jong Wook Kim1,2,7,8*, Christian Berrios2,4*, Miju Kim1,2*, Amy E. Schade2,4*, Guillaume Adelmant5, Huwate Yeerna7, Emily Damato1, Amanda Balboni Iniguez1,6, Selene K. Swanson9, Laurence Florens9, Michael P. Washburn9.10, Kim Stegmaier1,6, Nathaniel S. Gray5, Pablo Tamayo7,8, Ole Gjoerup2, Jarrod A. Marto5, James A. DeCaprio2,3,4,†, William C. Hahn1, 2, 3,† 1Broad Institute of Harvard and MIT, Cambridge MA. 2Department of Medical Oncology, Dana-Farber Cancer Institute, Boston MA. 3Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston MA. 4Program in Virology, Graduate School of Arts and Sciences, Harvard University, Cambridge, MA. 5Department of Cancer Biology and Blais Proteomics Center, Dana-Farber Cancer Institute, Boston MA. 6Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston MA. 7Division of Medical Genetics, School of Medicine, UC San Diego, CA. 8Moores Cancer Center, University of California, San Diego, CA. 9Stowers Institute for Medical Research, Kansas City, MO 64110 10Department of Pathology and Laboratory Medicine, University of Kansas Medical Center, Kansas City, KS 66160 * These authors contributed equally to the work. †Co-senior authors 1 bioRxiv preprint doi: https://doi.org/10.1101/823096; this version posted October 29, 2019. -

Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation
International Journal of Molecular Sciences Review Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation 1,2, 2, 3 2 3 Shu Wei y, Ye Li y, Sean P. Polster , Christopher R. Weber , Issam A. Awad and Le Shen 2,3,* 1 Graduate Program in Public Health and Preventive Medicine, Wuhan University of Science and Technology, Wuhan 430081, China; [email protected] 2 Department of Pathology, The University of Chicago, Chicago, IL 60615, USA; [email protected] (Y.L.); [email protected] (C.R.W.) 3 Section of Neurosurgery, Department of Surgery, The University of Chicago, Chicago, IL 60615, USA; [email protected] (S.P.P.); [email protected] (I.A.A.) * Correspondence: [email protected] These authors contributed equally. y Received: 10 December 2019; Accepted: 15 January 2020; Published: 20 January 2020 Abstract: Cerebral cavernous malformation (CCM) is a disease characterized by mulberry shaped clusters of dilated microvessels, primarily in the central nervous system. Such lesions can cause seizures, headaches, and stroke from brain bleeding. Loss-of-function germline and somatic mutations of a group of genes, called CCM genes, have been attributed to disease pathogenesis. In this review, we discuss the impact of CCM gene encoded proteins on cellular signaling, barrier function of endothelium and epithelium, and their contribution to CCM and potentially other diseases. Keywords: cerebral cavernous malformation; endothelial barrier; epithelial barrier; Rho; ROCK; MEKK3 1. Introduction One of the key functions of endothelial and epithelial cells is to create a barrier that separates different tissue compartments, and in the case of skin, epithelial cells separate body and outer environment.