Functional Divergence of Mammalian Tfap2a and Tfap2b Transcription Factors For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reconstruction of the Global Neural Crest Gene Regulatory Network in Vivo
Reconstruction of the global neural crest gene regulatory network in vivo Ruth M Williams1, Ivan Candido-Ferreira1, Emmanouela Repapi2, Daria Gavriouchkina1,4, Upeka Senanayake1, Jelena Telenius2,3, Stephen Taylor2, Jim Hughes2,3, and Tatjana Sauka-Spengler1,∗ Supplemental Material ∗Lead and corresponding author: Tatjana Sauka-Spengler ([email protected]) 1University of Oxford, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, Oxford, OX3 9DS, UK 2University of Oxford, MRC Centre for Computational Biology, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 3University of Oxford, MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Oxford, OX3 9DS, UK 4Present Address: Okinawa Institute of Science and Technology, Molecular Genetics Unit, Onna, 904-0495, Japan A 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R1_5-6ss) log2(R1_8-10ss) log2(R1_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.92 0 r=0.99 0 r=0.96 0 r=0.99 0 r=0.96 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R2_non-NC) log2(R2_5-6ss) log2(R3_5-6ss) log2(R2_8-10ss) log2(R3_8-10ss) 25 25 25 25 25 20 20 20 20 20 15 15 15 15 15 10 10 10 10 10 log2(R1_5-6ss) log2(R2_5-6ss) log2(R1_8-10ss) log2(R2_8-10ss) log2(R1_non-NC) 5 5 5 5 5 0 r=0.94 0 r=0.96 0 r=0.95 0 r=0.96 0 r=0.95 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 0 5 10 15 20 25 log2(R3_non-NC) log2(R4_5-6ss) log2(R3_5-6ss) log2(R4_8-10ss) log2(R3_8-10ss) -

Gene PMID WBS Locus ABR 26603386 AASDH 26603386
Supplementary material J Med Genet Gene PMID WBS Locus ABR 26603386 AASDH 26603386 ABCA1 21304579 ABCA13 26603386 ABCA3 25501393 ABCA7 25501393 ABCC1 25501393 ABCC3 25501393 ABCG1 25501393 ABHD10 21304579 ABHD11 25501393 yes ABHD2 25501393 ABHD5 21304579 ABLIM1 21304579;26603386 ACOT12 25501393 ACSF2,CHAD 26603386 ACSL4 21304579 ACSM3 26603386 ACTA2 25501393 ACTN1 26603386 ACTN3 26603386;25501393;25501393 ACTN4 21304579 ACTR1B 21304579 ACVR2A 21304579 ACY3 19897463 ACYP1 21304579 ADA 25501393 ADAM12 21304579 ADAM19 25501393 ADAM32 26603386 ADAMTS1 25501393 ADAMTS10 25501393 ADAMTS12 26603386 ADAMTS17 26603386 ADAMTS6 21304579 ADAMTS7 25501393 ADAMTSL1 21304579 ADAMTSL4 25501393 ADAMTSL5 25501393 ADCY3 25501393 ADK 21304579 ADRBK2 25501393 AEBP1 25501393 AES 25501393 AFAP1,LOC84740 26603386 AFAP1L2 26603386 AFG3L1 21304579 AGAP1 26603386 AGAP9 21304579 Codina-Sola M, et al. J Med Genet 2019; 56:801–808. doi: 10.1136/jmedgenet-2019-106080 Supplementary material J Med Genet AGBL5 21304579 AGPAT3 19897463;25501393 AGRN 25501393 AGXT2L2 25501393 AHCY 25501393 AHDC1 26603386 AHNAK 26603386 AHRR 26603386 AIDA 25501393 AIFM2 21304579 AIG1 21304579 AIP 21304579 AK5 21304579 AKAP1 25501393 AKAP6 21304579 AKNA 21304579 AKR1E2 26603386 AKR7A2 21304579 AKR7A3 26603386 AKR7L 26603386 AKT3 21304579 ALDH18A1 25501393;25501393 ALDH1A3 21304579 ALDH1B1 21304579 ALDH6A1 21304579 ALDOC 21304579 ALG10B 26603386 ALG13 21304579 ALKBH7 25501393 ALPK2 21304579 AMPH 21304579 ANG 21304579 ANGPTL2,RALGPS1 26603386 ANGPTL6 26603386 ANK2 21304579 ANKMY1 26603386 ANKMY2 -

Watsonjn2018.Pdf (1.780Mb)
UNIVERSITY OF CENTRAL OKLAHOMA Edmond, Oklahoma Department of Biology Investigating Differential Gene Expression in vivo of Cardiac Birth Defects in an Avian Model of Maternal Phenylketonuria A THESIS SUBMITTED TO THE GRADUATE FACULTY In partial fulfillment of the requirements For the degree of MASTER OF SCIENCE IN BIOLOGY By Jamie N. Watson Edmond, OK June 5, 2018 J. Watson/Dr. Nikki Seagraves ii J. Watson/Dr. Nikki Seagraves Acknowledgements It is difficult to articulate the amount of gratitude I have for the support and encouragement I have received throughout my master’s thesis. Many people have added value and support to my life during this time. I am thankful for the education, experience, and friendships I have gained at the University of Central Oklahoma. First, I would like to thank Dr. Nikki Seagraves for her mentorship and friendship. I lucked out when I met her. I have enjoyed working on this project and I am very thankful for her support. I would like thank Thomas Crane for his support and patience throughout my master’s degree. I would like to thank Dr. Shannon Conley for her continued mentorship and support. I would like to thank Liz Bullen and Dr. Eric Howard for their training and help on this project. I would like to thank Kristy Meyer for her friendship and help throughout graduate school. I would like to thank my committee members Dr. Robert Brennan and Dr. Lilian Chooback for their advisement on this project. Also, I would like to thank the biology faculty and staff. I would like to thank the Seagraves lab members: Jailene Canales, Kayley Pate, Mckayla Muse, Grace Thetford, Kody Harvey, Jordan Guffey, and Kayle Patatanian for their hard work and support. -
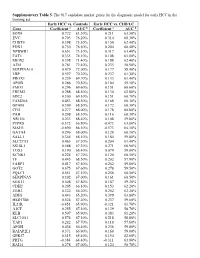
Supplementary Table 5. the 917 Candidate Marker Genes for the Diagnostic Model for Early HCC in the Training Set
Supplementary Table 5. The 917 candidate marker genes for the diagnostic model for early HCC in the training set. Early HCC vs. Controls Early HCC vs. CHB/LC Gene Coefficient a AUC b Coefficient a AUC b SOX9 0.722 81.30% 0.211 63.50% EVC 0.703 76.20% 0.314 68.30% CHST9 0.398 75.50% 0.150 62.40% PDX1 0.730 76.50% 0.204 60.40% NPBWR1 0.651 73.10% 0.317 63.40% FAT1 0.335 74.10% 0.108 61.00% MEIS2 0.398 71.40% 0.188 62.40% A2M 0.761 72.40% 0.235 58.90% SERPINA10 0.479 72.00% 0.177 58.40% LBP 0.597 70.20% 0.237 61.30% PROX1 0.239 69.70% 0.133 61.40% APOB 0.286 70.50% 0.104 59.10% FMO3 0.296 69.60% 0.151 60.60% FREM2 0.288 68.50% 0.130 62.80% SDC2 0.300 69.10% 0.151 60.70% FAM20A 0.453 68.50% 0.168 60.30% GPAM 0.309 68.50% 0.172 60.30% CFH 0.277 68.00% 0.178 60.80% PAH 0.208 68.30% 0.116 60.30% NR1H4 0.233 68.40% 0.108 59.80% PTPRS -0.572 66.80% -0.473 63.00% SIAH3 -0.690 66.10% -0.573 64.10% GATA4 0.296 68.00% 0.128 60.10% SALL1 0.344 68.10% 0.184 59.80% SLC27A5 0.463 67.30% 0.204 61.40% SS18L1 0.588 67.30% 0.271 60.90% TOX3 0.190 68.40% 0.079 59.00% KCNK1 0.224 67.70% 0.120 60.10% TF 0.445 68.50% 0.202 57.90% FARP1 0.417 67.50% 0.252 59.80% GOT2 0.675 67.60% 0.278 59.50% PQLC1 0.651 67.10% 0.258 60.50% SERPINA5 0.302 67.00% 0.161 60.50% SOX13 0.508 67.80% 0.187 59.30% CDH2 0.205 66.10% 0.153 62.20% ITIH2 0.322 66.20% 0.252 62.20% ADIG 0.443 65.20% 0.399 63.80% HSD17B6 0.524 67.20% 0.237 59.60% IL21R -0.451 65.90% -0.321 61.70% A1CF 0.255 67.10% 0.139 58.70% KLB 0.507 65.90% 0.383 61.20% SLC10A1 0.574 67.10% 0.218 58.80% YAP1 0.282 67.70% 0.118 -

SUPPLEMENTARY MATERIAL Bone Morphogenetic Protein 4 Promotes
www.intjdevbiol.com doi: 10.1387/ijdb.160040mk SUPPLEMENTARY MATERIAL corresponding to: Bone morphogenetic protein 4 promotes craniofacial neural crest induction from human pluripotent stem cells SUMIYO MIMURA, MIKA SUGA, KAORI OKADA, MASAKI KINEHARA, HIROKI NIKAWA and MIHO K. FURUE* *Address correspondence to: Miho Kusuda Furue. Laboratory of Stem Cell Cultures, National Institutes of Biomedical Innovation, Health and Nutrition, 7-6-8, Saito-Asagi, Ibaraki, Osaka 567-0085, Japan. Tel: 81-72-641-9819. Fax: 81-72-641-9812. E-mail: [email protected] Full text for this paper is available at: http://dx.doi.org/10.1387/ijdb.160040mk TABLE S1 PRIMER LIST FOR QRT-PCR Gene forward reverse AP2α AATTTCTCAACCGACAACATT ATCTGTTTTGTAGCCAGGAGC CDX2 CTGGAGCTGGAGAAGGAGTTTC ATTTTAACCTGCCTCTCAGAGAGC DLX1 AGTTTGCAGTTGCAGGCTTT CCCTGCTTCATCAGCTTCTT FOXD3 CAGCGGTTCGGCGGGAGG TGAGTGAGAGGTTGTGGCGGATG GAPDH CAAAGTTGTCATGGATGACC CCATGGAGAAGGCTGGGG MSX1 GGATCAGACTTCGGAGAGTGAACT GCCTTCCCTTTAACCCTCACA NANOG TGAACCTCAGCTACAAACAG TGGTGGTAGGAAGAGTAAAG OCT4 GACAGGGGGAGGGGAGGAGCTAGG CTTCCCTCCAACCAGTTGCCCCAAA PAX3 TTGCAATGGCCTCTCAC AGGGGAGAGCGCGTAATC PAX6 GTCCATCTTTGCTTGGGAAA TAGCCAGGTTGCGAAGAACT p75 TCATCCCTGTCTATTGCTCCA TGTTCTGCTTGCAGCTGTTC SOX9 AATGGAGCAGCGAAATCAAC CAGAGAGATTTAGCACACTGATC SOX10 GACCAGTACCCGCACCTG CGCTTGTCACTTTCGTTCAG Suppl. Fig. S1. Comparison of the gene expression profiles of the ES cells and the cells induced by NC and NC-B condition. Scatter plots compares the normalized expression of every gene on the array (refer to Table S3). The central line -

Genome-Wide Expression Analysis Ofptf1a- and Ascl1-Deficient Mice
The Journal of Neuroscience, April 24, 2013 • 33(17):7299–7307 • 7299 Development/Plasticity/Repair Genome-Wide Expression Analysis of Ptf1a- and Ascl1-Deficient Mice Reveals New Markers for Distinct Dorsal Horn Interneuron Populations Contributing to Nociceptive Reflex Plasticity Hendrik Wildner,1 Rebecca Das Gupta,2 Dominique Bro¨hl,3 Paul A. Heppenstall,4 Hanns Ulrich Zeilhofer,1,2* and Carmen Birchmeier3* 1Institute of Pharmacology and Toxicology, University of Zurich, CH-8057 Zurich, Switzerland, 2Institute of Pharmaceutical Sciences, Swiss Federal Institute of Technology (ETH) Zurich, CH-8093 Zurich, Switzerland, 3Department of Neuroscience, Max Delbru¨ck Center for Molecular Medicine, D-13125 Berlin, Germany, and 4Mouse Biology Unit, European Molecular Biology Laboratory, I-00015 Monterotondo, Italy Inhibitoryinterneuronsofthespinaldorsalhornplaycriticalrolesintheprocessingofnoxiousandinnocuoussensoryinformation.They form a family of morphologically and functionally diverse neurons that likely fall into distinct subtypes. Traditional classifications rely mainly on differences in dendritic tree morphology and firing patterns. Although useful, these markers are not comprehensive and cannot be used to drive specific genetic manipulations targeted at defined subsets of neurons. Here, we have used genome-wide expres- sion profiling of spinal dorsal horns of wild-type mice and of two strains of transcription factor-deficient mice (Ptf1aϪ/Ϫ and Ascl1/ Mash1Ϫ/Ϫ mice) to identify new genetic markers for specific subsets of dorsal horn inhibitory interneurons. Ptf1aϪ/Ϫ mice lack all inhibitory interneurons in the dorsal horn, whereas only the late-born inhibitory interneurons are missing in Ascl1Ϫ/Ϫ mice. We found 30 genes that were significantly downregulated in the dorsal horn of Ptf1aϪ/Ϫ mice. Twenty-one of those also showed reduced expression in Ascl1Ϫ/Ϫ mice. -

Single Cell Regulatory Landscape of the Mouse Kidney Highlights Cellular Differentiation Programs and Disease Targets
ARTICLE https://doi.org/10.1038/s41467-021-22266-1 OPEN Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets Zhen Miao 1,2,3,8, Michael S. Balzer 1,2,8, Ziyuan Ma 1,2,8, Hongbo Liu1,2, Junnan Wu 1,2, Rojesh Shrestha 1,2, Tamas Aranyi1,2, Amy Kwan4, Ayano Kondo 4, Marco Pontoglio 5, Junhyong Kim6, ✉ Mingyao Li 7, Klaus H. Kaestner2,4 & Katalin Susztak 1,2,4 1234567890():,; Determining the epigenetic program that generates unique cell types in the kidney is critical for understanding cell-type heterogeneity during tissue homeostasis and injury response. Here, we profile open chromatin and gene expression in developing and adult mouse kidneys at single cell resolution. We show critical reliance of gene expression on distal regulatory elements (enhancers). We reveal key cell type-specific transcription factors and major gene- regulatory circuits for kidney cells. Dynamic chromatin and expression changes during nephron progenitor differentiation demonstrates that podocyte commitment occurs early and is associated with sustained Foxl1 expression. Renal tubule cells follow a more complex differentiation, where Hfn4a is associated with proximal and Tfap2b with distal fate. Mapping single nucleotide variants associated with human kidney disease implicates critical cell types, developmental stages, genes, and regulatory mechanisms. The single cell multi-omics atlas reveals key chromatin remodeling events and gene expression dynamics associated with kidney development. 1 Renal, Electrolyte, and Hypertension Division, Department of Medicine, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA. 2 Institute for Diabetes, Obesity, and Metabolism, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks
bioRxiv preprint doi: https://doi.org/10.1101/219667; this version posted November 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Article Modeling Enhancer-Promoter Interactions with Attention-Based Neural Networks Weiguang Mao 1,2, Dennis Kostka 1,2,3 and Maria Chikina 1,2* 1 Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh 2 Joint Carnegie Mellon - University of Pittsburgh Ph.D. Program in Computational Biology 3 Department of Developmental Biology, School of Medicine, University of Pittsburgh * Correspondence: [email protected] Academic Editor: name Version November 9, 2017 submitted to Cells 1 Abstract: Background: Gene regulatory sequences play critical roles in ensuring tightly controlled 2 RNA expression patterns that are essential in a large variety of biological processes. Specifically, 3 enhancer sequences drive expression of their target genes, and the availability of genome-wide 4 maps of enhancer-promoter interactions has opened up the possibility to use machine learning 5 approaches to extract and interpret features that define these interactions in different biological 6 contexts. Methods: Inspired by machine translation models we develop an attention-based neural 7 network model, EPIANN, to predict enhancer-promoter interactions based on DNA sequences. Codes 8 and data are available at https://github.com/wgmao/EPIANN. Results: Our approach accurately 9 predicts enhancer-promoter interactions across six cell lines. In addition, our method generates 10 pairwise attention scores at the sequence level, which specify how short regions in the enhancer and 11 promoter pair-up to drive the interaction prediction. -

Genetic Factors in Nonsyndromic Orofacial Clefts
Published online: 2021-02-12 THIEME Review Article 101 Genetic Factors in Nonsyndromic Orofacial Clefts Mahamad Irfanulla Khan1 Prashanth C.S.2 Narasimha Murthy Srinath3 1 Department of Orthodontics & Dentofacial Orthopedics, The Oxford Address for correspondence Mahamad Irfanulla Khan, BDS, MDS, Dental College, Bangalore, Karnataka, India Department of Orthodontics & Dentofacial Orthopedics, The Oxford 2 Department of Orthodontics & Dentofacial Orthopedics, DAPM R.V. Dental College, Bangalore, Karnataka 560068, India Dental College, Bangalore, Karnataka, India (e-mail: [email protected]). 3 Department of Oral & Maxillofacial Surgery, Krishnadevaraya College of Dental Sciences, Bangalore, Karnataka, India Global Med Genet 2020;7:101–108. Abstract Orofacial clefts (OFCs) are the most common congenital birth defects in humans and immediately recognized at birth. The etiology remains complex and poorly understood and seems to result from multiple genetic and environmental factors along with gene– environment interactions. It can be classified into syndromic (30%) and nonsyndromic (70%) clefts. Nonsyndromic OFCs include clefts without any additional physical or Keywords cognitive deficits. Recently, various genetic approaches, such as genome-wide associ- ► orofacial clefts ation studies (GWAS), candidate gene association studies, and linkage analysis, have ► nonsyndromic identified multiple genes involved in the etiology of OFCs. ► genetics This article provides an insight into the multiple genes involved in the etiology of OFCs. ► gene mutation Identification of specific genetic causes of clefts helps in a better understanding of the ► genome-wide molecular pathogenesis of OFC. In the near future, it helps to provide a more accurate association study diagnosis, genetic counseling, personalized medicine for better clinical care, and ► linkage analysis prevention of OFCs. -

Upregulation of the Transcription Factor TFAP2D Is Associated With
Fraune et al. Molecular Medicine (2020) 26:24 Molecular Medicine https://doi.org/10.1186/s10020-020-00148-4 RESEARCH ARTICLE Open Access Upregulation of the transcription factor TFAP2D is associated with aggressive tumor phenotype in prostate cancer lacking the TMPRSS2:ERG fusion Christoph Fraune1†, Luisa Harms1†, Franziska Büscheck1, Doris Höflmayer1, Maria Christina Tsourlakis1, Till S. Clauditz1, Ronald Simon1* , Katharina Möller1, Andreas M. Luebke1, Christina Möller-Koop1, Stefan Steurer1, Claudia Hube-Magg1, Guido Sauter1, Sören Weidemann1, Patrick Lebok1, David Dum1, Simon Kind1, Sarah Minner1, Jakob R. Izbicki2, Thorsten Schlomm3, Hartwig Huland4, Hans Heinzer4, Eike Burandt1, Alexander Haese4, Markus Graefen4 and Cornelia Schroeder2 Abstract Background: TFAP2D is a transcription factor important for modulating gene expression in embryogenesis. Its expression and prognostic role in prostate cancer has not been evaluated. Methods: Therefore, a tissue microarray containing 17,747 prostate cancer specimens with associated pathological, clinical, and molecular data was analyzed by immunohistochemistry to assess the role of TFAP2D. Results: TFAP2D expression was typically increased in prostate cancer as compared to adjacent non-neoplastic glands. TFAP2D staining was considered negative in 24.3% and positive in 75.7% of 13,545 interpretable cancers. TFAP2D staining was significantly linked to advanced tumor stage, high classical and quantitative Gleason grade, lymph node metastasis, and a positive surgical margin (p ≤ 0.0045). TFAP2D positivity was more common in ERG fusion positive (88.7%) than in ERG negative cancers (66.8%; p < 0.0001). Subset analyses in 3776 cancers with and 4722 cancers without TMPRSS2:ERG fusion revealed that associations with tumor phenotype and patient outcome were largely driven by the subset of ERG negative tumors.