The Genomics of Emerging Pathogens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GB Virus C/Hepatitis G Virus Does Not Induce Expression of P44 Antigen in Chimpanzee Hepatocytes Yohko K
Jpn. J. Infect. Dis., 54, 2001 and skin were removed was purchased on the previous night ongln Of infection was not clear. The food service in a festival of the festival. It was left in the room temperature ovemight. as described in the present case does not requlre regulation The cooking started at 4 0'clock in the momlng by roastlng under the food hyglene law. This incident indicated the the bulk meat by rotation over a gas bumer. The meat was necessity of proper measures for food security in this type of covered by a large steel box during roastlng. It was cut into short temporary food service and regulatory measures in case small pleCeS and served at 10 o'clock in the momlng. Some of possible occurrences of food poISOnlng. noted that some portion of the meat was rare; i.e., it was insufficiently cooked. Laboratory and other epidemiologlCal data will be published ln the outbreak, a total 58 cases were counted. There were in Infectious Agents Surveillance Report, vol. 22 (June, 200 I ). 41 primary infections, 1 1 secondary infections including a We thank the clinical institutions, schools and other institu- case which took place in a nursery school, and 6 cases whose tions fわr their collaboration. Laboratory and Epidemiology Communications GB Virus C/Hepatitis G Virus Does Not Induce Expression of p44 Antigen in Chimpanzee Hepatocytes Yohko K. Shimizu*, Minako Hijikata and Hiroshi Yoshikural Department ofRespiratoyy Diseases, Research Institute, International Medical Center ofJapan, Toyama 1-21-1 Shinjuku-ku, Too,0 162-8655 and JNational Institute ofInfectious Diseases, Toyama 1-23-1, Shinjuku-ku, Tokyo 162-8640 Communicated by Hiroshi Ybshikura (Accepted June 1, 2001) The cytoplasmic antlgen, p44, was orlglnally discovered found chimpanzees whose sera were positive for GBV-C什IGV in hepatocytes of chimpanzees experimentally infected with RNA by RTIPCR. -

Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients
Jpn. J. Infect. Dis., 59, 25-30, 2006 Original Article Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients with Chronic Hepatitis B or C Jeng-Fu Yang1,2, Chia-Yen Dai2,3, Wan-Long Chuang2, Wen-Yi Lin1,2, Zu-Yau Lin2, Shinn-Cherng Chen2, Ming-Yuh Hsieh2, Liang-Yen Wang2, Jung-Fa Tsai2, Wen-Yu Chang2 and Ming-Lung Yu1,2* 1Department of Preventive Medicine and 2Department of Medicine, Kaohsiung Medical University Hospital, and 3Department of Occupational Medicine, Kaohsiung Municipal HsiaoKang Hospital, Kaohsiung, Taiwan (Received July 5, 2005. Accepted November 11, 2005) SUMMARY: Hepatitis G virus/GB virus-C (HGV/GBV-C) is a newly identified Flavivirus. Its clinical signifi- cance in chronic hepatitis B and C remains controversial. Infection with HGV/GBV-C was surveyed in 500 blood donors, 130 patients with chronic hepatitis B and 173 with hepatitis C, with chronic liver disease, cirrhosis, and/or hepatocellular carcinoma (HCC). HGV/GBV-C RNA was detected by reverse transcription-polymerase chain reaction. An antibody to HGV/GBV-C’s second envelope protein (anti-E2 Ab) was detected using an enzyme immunoassay. The prevalence of HGV/GBV-C RNA was 3.4% and the exposure rate 10.2% in blood donors. The prevalence of HGV/GBV-C RNA in patients with chronic hepatitis B and hepatitis C was 7.7 and 17.3%, respectively (P = 0.002). The prevalence of the HGV/GBV-C infection in hepatitis B carriers increased with the severity of chronic liver disease and risk of HCC. The age and duration of hepatitis B virus infection were the more important contributing factors. -

Handbook for Parents
Contributors Cassandra L. Aspinall, MSW, LICSW Christine Feick, MSW Craniofacial Center, Seattle Children’s Hospital; Ann Arbor, MI University of Washington School of Social Work, Seattle, WA Sallie Foley, LMSW Certified Sex Therapist, AASECT; Dept. Social Arlene B. Baratz, MD Work/Sexual Health, University of Michigan Medical Advisor, Androgen Insensitivity Health Systems, Ann Arbor, MI Syndrome Support Group, Pittsburgh, PA Joel Frader, MD, MA Max & Tamara Beck General Academic Pediatrics, Children’s Atlanta, GA Memorial Hospital; Dept. Pediatrics and Program in Medical Humanities & Bioethics, William Byne, MD Feinberg School of Medicine, Northwestern Psychiatry, Mount Sinai Medical Center, New University, Chicago, IL York, NY Jane Goto David Cameron Board of Directors, Intersex Society of North Intersex Society of North America, San America; Board of Directors, Androgen Francisco, CA Insensitivity Syndrome Support Group, Seattle, Anita J. Catlin, DSNc, FNP, FAAN WA Nursing and Ethics, Sonoma State University, Michael Grant Sonoma, CA Lansing, MI Cheryl Chase Janet Green Founder and Executive Director, Intersex Society Co-Founder, Bodies Like Ours; Board of of North America, Rohnert Park, CA Directors, CARES Foundation; Board of Kimberly Chu, LCSW, DCSW Overseers, Beth Israel Hospital; Board of Department of Child & Adolescent Psychiatry, Trustees, Continuum Healthcare, New York, Mount Sinai Medical Center, New York, NY NY Howard Devore Philip A. Gruppuso, MD San Francisco, CA Associate Dean of Medical Education, Brown University; Pediatric Endocrinology, Rhode Alice Dreger, Ph.D. (Project Coordinator and Island Hospital, Providence, RI Editor) Program in Medical Humanities and Bioethics, William G. Hanley, BPS Feinberg School of Medicine, Northwestern Memphis, TN University, Chicago, IL iii iv Debora Rode Hartman Charmian A. -
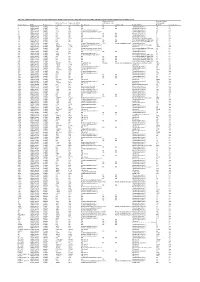
Table S4. Comparison Between the Discvr Results and the BLAST
Table S4. Comparison between the DisCVR results and the BLAST results from the original manuscript for healthy patients and patients with unexplained acute febrile illness Number of k-mers Number of reads Number of k-mers Number of distinct matching the 2nd matching with Sample_Name RUN Patient status matching k-mers matching Top Virus hits hit 2nd Virus Hit Top BLAST hit BLAST Concordance 3 SRR1748140 healthy NA NA NA NA NA Human.adenovirus.C 10 no 4 SRR1748141 healthy NA NA NA NA NA Human.adenovirus.C 65 no 8 SRR1748143 healthy 943 379 Human mastadenovirus C Human.adenovirus.C 65 yes 10 SRR1748147 healthy 1669 133 Human immunodeficiency virus 1 Human.adenovirus.C 26 no 14 SRR1748151 healthy NA NA NA NA NA Human.adenovirus.C 4 no 15 SRR1748152 healthy NA NA NA NA NA Human.adenovirus.C 6 no 108 SRR1748162 healthy NA NA NA NA NA Human.adenovirus.C 6 no 137 SRR1748172 healthy 15343 4714 Human immunodeficiency virus 1 Human.immunodeficiency.virus 314 yes 173 SRR1748173 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 20 no 245 SRR1748177 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 17 no 312 SRR1748178 healthy 2843 4 Human T-lymphotropic virus 1 1282 Human mastadenovirus C Human.adenovirus.C 70 no 316 SRR1748179 healthy 22915 5941 Human immunodeficiency virus 1 Human.immunodeficiency.virus 342 yes 319 SRR1748180 healthy 2247683 21201 GB virus C GB.virus.C 20629 yes 325 SRR1748181 healthy 1006 177 Human immunodeficiency virus 1 Heterosigma.akashiwo.RNA.virus 60 no 329 SRR1748182 healthy 1513 124 Human immunodeficiency virus 1 -

Genetics, Underlying Pathologies and Psychosexual Differentiation Valerie A
REVIEWS DSDs: genetics, underlying pathologies and psychosexual differentiation Valerie A. Arboleda, David E. Sandberg and Eric Vilain Abstract | Mammalian sex determination is the unique process whereby a single organ, the bipotential gonad, undergoes a developmental switch that promotes its differentiation into either a testis or an ovary. Disruptions of this complex genetic process during human development can manifest as disorders of sex development (DSDs). Sex development can be divided into two distinct processes: sex determination, in which the bipotential gonads form either testes or ovaries, and sex differentiation, in which the fully formed testes or ovaries secrete local and hormonal factors to drive differentiation of internal and external genitals, as well as extragonadal tissues such as the brain. DSDs can arise from a number of genetic lesions, which manifest as a spectrum of gonadal (gonadal dysgenesis to ovotestis) and genital (mild hypospadias or clitoromegaly to ambiguous genitalia) phenotypes. The physical attributes and medical implications associated with DSDs confront families of affected newborns with decisions, such as gender of rearing or genital surgery, and additional concerns, such as uncertainty over the child’s psychosexual development and personal wishes later in life. In this Review, we discuss the underlying genetics of human sex determination and focus on emerging data, genetic classification of DSDs and other considerations that surround gender development and identity in individuals with DSDs. Arboleda, V. A. et al. Nat. Rev. Endocrinol. advance online publication 5 August 2014; doi:10.1038/nrendo.2014.130 Introduction Sex development is a critical component of mammalian disrupted, which occurs primarily as a result of genetic development that provides a robust mechanism for con- mutations that interfere with either the development of tinued generation of genetic diversity within a species. -

GB/Hepatitis G Viruses Likelihood of Secondary Transmission: • Probably Frequent, Based on the Prevalence of Virus in Disease Agent: Blood Donors
APPENDIX 2 GB/Hepatitis G Viruses Likelihood of Secondary Transmission: • Probably frequent, based on the prevalence of virus in Disease Agent: blood donors. • GB viruses (GBV-C/HGV is an acronym for GB virus-C At-Risk Populations: and hepatitis G virus strain variants.) • Blood recipients, injection-drug users, and infants Disease Agent Characteristics: born to infected mothers • Family: Flaviviridae; Genus: Unclassified for GBV-C Vector and Reservoir Involved: • Virion morphology and size: Enveloped, nucleo- capsid of unknown symmetry, 50-100 nm in diameter • Humans • Nucleic acid: Linear, positive-sense, single-stranded Blood Phase: RNA, ~9.4 kb in length • Physicochemical properties: Less stable in CsCl than • Viremic phase can last from weeks to years HCV; other properties not established for these Survival/Persistence in Blood Products: viruses, but, under in vitro conditions, other flavivi- ruses are stable in alkaline environment of pH 8 and • Survives refrigeration are sensitive to treatment with heat, organic solvents, • Inactivated by solvent-detergent and detergents. Transmission by Blood Transfusion: Disease Name: • Well documented in prospective studies • No known disease association Cases/Frequency in Population: Priority Level: • The prevalence of viremia is 1-4%, and antibody • Scientific/Epidemiologic evidence regarding blood prevalence is 3-14% in blood donors. safety: Absent; transmission documented, but no • Prevalence of 10-20% in patients with viral and non- disease associated despite extensive studies viral liver diseases based on antibody and RNA • Public perception and/or regulatory concern regard- • Prevalence of 75-90% in injection-drug users (anti- ing blood safety: Absent body and RNA) • Public concern regarding disease agent: Absent Incubation Period: Background: • Viremia becomes detectable from 2 days to 2 weeks • In the 1960s, serum from a surgeon (GB) with acute postexposure. -

8.3 Notebook Annotated
Hans L. Tillmann, md Department of Gastroenterology, When Two Infections Hepatology, and Endocrinology Diagnostic Laboratory for Viral and Autoimmune Hepatitis Are Better Than One: Medizinische Hochshule Hannover, Germany The Exceptional Role of Summary by Tim Horn Edited by Douglas Dieterich, md, and GB Virus-C (GBV-C) in Stephen Locarnini, mbbs, phd, frc (path) HIV Disease Reprinted from The prn Notebook® | september 2003 | Dr. James F. Braun, Editor-in-Chief | Tim Horn, Executive Editor. | Published in New York City by the Physicians’ Research Network, Inc.® John Graham Brown, Executive Director | For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©september 2003 when it comes to hiv and hepatitis virus coinfections, the pages fected with hcv (Linnen, 1996). A second virus was isolated in this pa- of The prn Notebook have been filled with numerous reports high- tient, which the Genelabs team called the hepatitis G virus (hgv). lighting the distressing prevalence and negative consequences of both With the discovery of both viruses, researchers set out to deter- hepatitis B virus (hbv) and hepatitis C virus (hcv) in hiv-infected in- mine the full genomic sequences of the isolates. It turned out that dividuals. However, it does not appear that all coinfections are harmful. gbv-c and hgv are closely related isolates of the same virus, with more In fact, some may be associated with a significant survival advantage— than 95% sequence homology (Alter, 1996). It was also determined a theory that has many hiv experts both perplexed and excited. that the genomic organization of these viruses is similar to that of hcv gb virus-c (gbv-c) infection is one such example. -

Lessons Learned on Data Sharing in COVID-19 Pandemic Can Inform Future Outbreak Preparedness and Response” Science & Diplomacy, Vol
Jonathan LoTempio, D’Andre Spencer, Rebecca Yarvitz, Arthur Delot Vilain, Eric Vilain, and Emmanuèle Délot, “We Can Do Better: Lessons Learned on Data Sharing in COVID-19 Pandemic Can Inform Future Outbreak Preparedness and Response” Science & Diplomacy, Vol. 9, No. 2 (June 2020). https://www.sciencediplomacy.org/article/2020/we-can-do-better- lessons-learned-data-sharing-in-covid-19-pandemic-can-inform-future This copy is for non-commercial use only. More articles, perspectives, editorials, and letters can be found at www.sciencediplomacy.org. Science & Diplomacy is published by the Center for Science Diplomacy of the American Association for the Advancement of Science (AAAS), the world’s largest general scientific society. We Can Do Better: Lessons Learned on Data Sharing in COVID-19 Pandemic Can Inform Future Outbreak Preparedness and Response Jonathan LoTempio, D’Andre Spencer, Rebecca Yarvitz, Arthur Delot Vilain, Eric Vilain, and Emmanuèle Délot he COVID-19 pandemic will remain a critical issue until a safe and Teffective vaccine is in global use. A strong international network exists for the systematic collection and sharing of influenza genome sequence data, which has proven extensible to COVID-19.¹ However, the robust demographic and clinical data needed to understand the progression of COVID-19 within individuals and across populations are collected by an array of local, regional, federal, and/or national agencies, with country-specific, often overlapping mandates. The networks tasked with transmitting descriptive, disaggregated data have not done so in a standardized manner; most data are made available in variable, incompatible forms, and there is no central, global hub. -

Tissue-Specific Expression and Regulation of Sexually Dimorphic Genes in Mice
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Tissue-specific expression and regulation of sexually dimorphic genes in mice Xia Yang,1 Eric E. Schadt,2 Susanna Wang,3 Hui Wang,4 Arthur P. Arnold,5 Leslie Ingram-Drake,3 Thomas A. Drake,6 and Aldons J. Lusis1,3,7 1Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, California 90095, USA; 2Rosetta Inpharmatics, LLC, a Wholly Owned Subsidiary of Merck & Co. Inc., Seattle, Washington 98109, USA; 3Department of Human Genetics, University of California, Los Angeles, California 90095, USA; 4Department of Statistics, College of Letters and Science, University of California, Los Angeles, California 90095, USA; 5Department of Physiological Science, and Laboratory of Neuroendocrinology of the Brain Research Institute, University of California, Los Angeles, California 90095, USA; 6Department of Pathology and Laboratory Medicine, University of California, Los Angeles, California 90095, USA We report a comprehensive analysis of gene expression differences between sexes in multiple somatic tissues of 334 mice derived from an intercross between inbred mouse strains C57BL/6J and C3H/HeJ. The analysis of a large number of individuals provided the power to detect relatively small differences in expression between sexes, and the use of an intercross allowed analysis of the genetic control of sexually dimorphic gene expression. Microarray analysis of 23,574 transcripts revealed that the extent of sexual dimorphism in gene expression was much greater than previously recognized. Thus, thousands of genes showed sexual dimorphism in liver, adipose, and muscle, and hundreds of genes were sexually dimorphic in brain. -

The Breadth of Viruses in Human Semen
Article DOI: https://doi.org/10.3201/eid2311.171049 The Breadth of Viruses in Human Semen Technical Appendix Technical Appendix Table. Viruses that are capable of causing viremia and found in human semen* Detection in semen, Isolation from semen Evidence for sexual maximum detection maximum detection transmission within same Virus Family time, d time, d cohort Adenoviruses Adenoviridae AD (1) RCC Unknown Transfusion transmitted virus Anelloviridae NAA (2) No data found Unknown Lassa fever virus† Arenaviridae NAA, 103 (3) RCC, 20 (3) Unknown Rift Valley fever virus† Bunyaviridae NAA, 117 (4) No data found Unknown Ebola virus Filoviridae NAA, 531 (5) RCC, 82 (6) Epi + mol + sem (7) Marburg virus† Filoviridae AD, 83 (8) RAS, 83 (8) Epi + sem (9) GB virus C Flaviviridae NAA (10) No data found Epi + mol (11) Hepatitis C virus Flaviviridae NAA (12); AD (13) No data found Epi + mol (14) Zika virus Flaviviridae NAA, 188 (15) RCC, 7 (16) Epi + mol + sem (17) Hepatitis B virus Hepadnaviridae NAA (18); AD (19) RAS (20) Epi + mol (21) Cytomegalovirus Herpesviridae NAA (22) RCC (23) Epi + mol + sem (24) Epstein Barr virus Herpesviridae NAA (22) No data found Epi and semen (25) Human herpes virus 8 Herpesviridae NAA (26) RCC (27) Epi + mol (28) Human herpes virus 7 Herpesviridae NAA (29) No data found Unknown Human herpes virus 6 Herpesviridae NAA (22) No data found Unknown Human simplex viruses 1 and 2 Herpesviridae NAA (22); AD (1) RCC (1) Epi + mol + sem (30) Varicella zoster virus Herpesviridae NAA (22) No data found Unknown Mumps virus† Paramyxoviridae -

DSD Symposium
DSD Symposium October 13-14 2006 Renaissance Parc 55 Hotel, San Francisco Presented by Intersex Society of North America and Gay and Lesbian Medical Association Contents Agenda for Change: Psychology and Clinical Management of Disorders of Sex Development in Adulthood Lih-Mei Liao PhD Report on Chicago Consensus Conference David Sandberg PhD, Cheryl Chase, and Eric Vilain PhD MD Nomenclature Change: I Am Not a Disorder Katie Baratz, Arlene Baratz MD, Eric Vilain PhD MD, and Peter Trinkl Counseling Adults William Byne MD and Nina Williams PhD How to Build a Team Barbara Neilson PhD and Melissa Parisi PhD Handbook for Parents Arlene Baratz MD DSDs and Cancer: Caring for Intersex Patients Katie Baratz Counseling Parents David Sandberg PhD, Arlene Baratz MD, and Ellen Feder PhD Setting the Research Agenda Lih-Mei Liao PhD DSD Symposium Parc 55 Hotel San Francisco October 13-14 2006 www.isna.org Introduction The Intersex Society of North America hosted the First DSD Symposium, a gathering of intersex adults, parents, and allied healthcare professionals, October 13-14, 2006, at the Renaissance Parc 55 Hotel in San Francisco. This was a chance to meet and learn from others working to improve the quality of healthcare for families with children born with Disorders of Sex Development, and for adults dealing with the many ongoing healthcare concerns that result from DSDs. The DSD Symposium was a mini-conference, held within the Gay and Lesbian Medical Association’s annual conference. Registrants to the GLMA conference (about 400 people) were welcome to attend all DSD Symposium presentations. There were about 40 people registered for the DSD Symposium alone. -

Download the Background and Timeline Insert
HUMAN “They’re Chasing Us Away RIGHTS from Sport” WATCH Human Rights Violations in Sex Testing of Elite Women Athletes 19301930ss 19401940s-1950s-1950s s Unsubstantiated stories of men Systematic sex testing, of a sort, exists as early as masquerading as women in international the 1940s via identity cards and “certificates of sporting events first appear.i femininity,” with the IAAF and IOC requiring all female athletes who wish to register for an event to provide a physician letter attesting to their sex for eligibility purposes.ii Meeting of the Executive Committee of the International Olympic Committee in 1951 in Vienna, chaired by IOC President Edström. © 1951 ullstein bild via Getty Images BACKGROUND You have to sacrifice so much, especially us ladies. —C.M., athlete, November 12, 2019 The regulation of women’s participation in sport via “sex testing” dates back decades. A key architect of such regulations—a former official with the International Olympic Committee (IOC) and World Athletics—later went on to characterize previous testing regimes as a “systematic violation for which the world of sport must take respon- sibility,” and “a decades-long example of sexual harassment of sexual abuse within sport [and] a flagrant abuse.”13 The earliest attempts at “sex testing” that sports authorities instituted in the 1940s for the purposes of eligibility were informal and ad hoc, but by the 1960s, sports governing bodies such as the IAAF and the IOC began system- atic mandatory testing of all women athletes based on rumors that some women “were more male than female,” resulting in “unfair competition for ‘real’ women.”14 There have never been analogous regulations for men.