PATHOLOGY of HIV/AIDS 32Nd Edition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Male Reproductive System
Management of Men’s Reproductive 3 Health Problems Men’s Reproductive Health Curriculum Management of Men’s Reproductive 3 Health Problems © 2003 EngenderHealth. All rights reserved. 440 Ninth Avenue New York, NY 10001 U.S.A. Telephone: 212-561-8000 Fax: 212-561-8067 e-mail: [email protected] www.engenderhealth.org This publication was made possible, in part, through support provided by the Office of Population, U.S. Agency for International Development (USAID), under the terms of cooperative agreement HRN-A-00-98-00042-00. The opinions expressed herein are those of the publisher and do not necessarily reflect the views of USAID. Cover design: Virginia Taddoni ISBN 1-885063-45-8 Printed in the United States of America. Printed on recycled paper. Library of Congress Cataloging-in-Publication Data Men’s reproductive health curriculum : management of men’s reproductive health problems. p. ; cm. Companion v. to: Introduction to men’s reproductive health services, and: Counseling and communicating with men. Includes bibliographical references. ISBN 1-885063-45-8 1. Andrology. 2. Human reproduction. 3. Generative organs, Male--Diseases--Treatment. I. EngenderHealth (Firm) II. Counseling and communicating with men. III. Title: Introduction to men’s reproductive health services. [DNLM: 1. Genital Diseases, Male. 2. Physical Examination--methods. 3. Reproductive Health Services. WJ 700 M5483 2003] QP253.M465 2003 616.6’5--dc22 2003063056 Contents Acknowledgments v Introduction vii 1 Disorders of the Male Reproductive System 1.1 The Male -

A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for High-Grade Anal Intraepithelial Neoplasia
Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for High-Grade Anal Intraepithelial Neoplasia Using The BARRX™ Anorectal Wand (Clinical Protocol 01-2017) AUGUST 31, 2017 Page 1 of 40 Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia PROTOCOL SIGNATURE PAGE I, , Principal Investigator at site , agree to conduct and follow this protocol: PROTOCOL 01-2017: A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for Anal Intraepithelial Neoplasia using the Barrx™ Anorectal Wand as written according to FDA guidelines. I understand that no deviations from the above protocol may be made without written permission from the Protocol Chair(s). _________________________________ _____________________ Signature Date (mm/dd/yyyy) Page 2 of 40 Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia P ROT O COL S U M M ARY Title A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for Anal Intraepithelial Neoplasia using the Barrx™ Anorectal Wand Design Multi-center prospective trial involving up to 70 subjects Subject Population HIV-positive and HIV-negative subjects with intra-anal intraepithelial neoplasia (AIN) containing at least one high- grade squamous intraepithelial lesions (HSIL) involving the squamocolumnar junction (SCJ). Objective Assess the safety, and efficacy of circumferential radiofrequency ablation (RFA) to the anal canal using the FDA cleared Barrx™ Anorectal Wand to eradicate anal -

USMLE – What's It
Purpose of this handout Congratulations on making it to Year 2 of medical school! You are that much closer to having your Doctor of Medicine degree. If you want to PRACTICE medicine, however, you have to be licensed, and in order to be licensed you must first pass all four United States Medical Licensing Exams. This book is intended as a starting point in your preparation for getting past the first hurdle, Step 1. It contains study tips, suggestions, resources, and advice. Please remember, however, that no single approach to studying is right for everyone. USMLE – What is it for? In order to become a licensed physician in the United States, individuals must pass a series of examinations conducted by the National Board of Medical Examiners (NBME). These examinations are the United States Medical Licensing Examinations, or USMLE. Currently there are four separate exams which must be passed in order to be eligible for medical licensure: Step 1, usually taken after the completion of the second year of medical school; Step 2 Clinical Knowledge (CK), this is usually taken by December 31st of Year 4 Step 2 Clinical Skills (CS), this is usually be taken by December 31st of Year 4 Step 3, typically taken during the first (intern) year of post graduate training. Requirements other than passing all of the above mentioned steps for licensure in each state are set by each state’s medical licensing board. For example, each state board determines the maximum number of times that a person may take each Step exam and still remain eligible for licensure. -

Q Fever in Small Ruminants and Its Public Health Importance
Journal of Dairy & Veterinary Sciences ISSN: 2573-2196 Review Article Dairy and Vet Sci J Volume 9 Issue 1 - January 2019 Copyright © All rights are reserved by Tolera Tagesu Tucho DOI: 10.19080/JDVS.2019.09.555752 Q Fever in Small Ruminants and its Public Health Importance Tolera Tagesu* School of Veterinary Medicine, Jimma University, Ethiopia Submission: December 01, 2018; Published: January 11, 2019 *Corresponding author: Tolera Tagesu Tucho, School of Veterinary Medicine, Jimma University, Jimma Oromia, Ethiopia Abstract Query fever is caused by Coxiella burnetii, it’s a worldwide zoonotic infectious disease where domestic small ruminants are the main reservoirs for human infections. Coxiella burnetii, is a Gram-negative obligate intracellular bacterium, adapted to thrive within the phagolysosome of the phagocyte. Humans become infected primarily by inhaling aerosols that are contaminated with C. burnetii. Ingestion (particularly drinking raw milk) and person-to-person transmission are minor routes. Animals shed the bacterium in urine and feces, and in very high concentrations in birth by-products. The bacterium persists in the environment in a resistant spore-like form which may become airborne and transported long distances by the wind. It is considered primarily as occupational disease of workers in close contact with farm animals or processing their be commenced immediately whenever Q fever is suspected. To prevent both the introduction and spread of Q fever infection, preventive measures shouldproducts, be however,implemented it may including occur also immunization in persons without with currently direct contact. available Doxycycline vaccines drugof domestic is the first small line ruminant of treatment animals for Q and fever. -
![Clinical Reviewer: [Joohee Lee ] STN: [125508/153 ]](https://docslib.b-cdn.net/cover/7770/clinical-reviewer-joohee-lee-stn-125508-153-127770.webp)
Clinical Reviewer: [Joohee Lee ] STN: [125508/153 ]
Clinical Reviewer: [Joohee Lee ] STN: [125508/153 ] Application sBLA Type STN 125508/153 CBER February 1, 2016 Received Date PDUFA Goal December 1, 2016 Date Division / DVRPA /OVRR Office Priority Review No Reviewer Joohee Lee Name(s) Review October 6, 2016 Completion Date / Stamped Date Supervisory Concurrence Lucia Lee, M.D., Team Leader, CRB1 Jeff Roberts, M.D., Chief, CRB1 Applicant Merck Established Human Papillomavirus 9-valent Vaccine, Recombinant Name (Proposed) Gardasil 9 Trade Name Pharmacologic Vaccine Class Page i Clinical Reviewer: [Joohee Lee ] STN: [125508/153 ] Formulation(s), A 0.5 mL dose contains: including Recombinant virus-like particles (VLPs) of the major capsid (L1) Adjuvants, etc protein of HPV types 6, 11, 16, 18, 31, 33, 45, 52, and 58)* adsorbed on preformed aluminum-containing adjuvant (Amorphous Aluminum Hydroxyphosphate Sulfate or AAHS) *Amounts of HPV type L1 protein are as follows: 30 mcg/40mcg/60mcg/40mcg/20mcg/20mcg/20mcg/20mcg/20mcg, respectively. Dosage 0.5-mL suspension for intramuscular injection as a single-dose Form(s) and vial and prefilled syringe Route(s) of Administration Dosing Two-dose regimen: 0 and 6 to 12 months Regimen A 3- dose regimen (0, 2 and 6 months) is approved for use in individuals 9 through 26 years of age (STN 125508/0) Page ii Clinical Reviewer: [Joohee Lee ] STN: [125508/153 ] Indication(s) and Intended This supplement introduces a 2-dose regimen in addition to the Population(s) licensed 3-dose regimen. The intended population for the 2- dose regimen is girls and boys 9 through 14 years of age. -

GB Virus C/Hepatitis G Virus Does Not Induce Expression of P44 Antigen in Chimpanzee Hepatocytes Yohko K
Jpn. J. Infect. Dis., 54, 2001 and skin were removed was purchased on the previous night ongln Of infection was not clear. The food service in a festival of the festival. It was left in the room temperature ovemight. as described in the present case does not requlre regulation The cooking started at 4 0'clock in the momlng by roastlng under the food hyglene law. This incident indicated the the bulk meat by rotation over a gas bumer. The meat was necessity of proper measures for food security in this type of covered by a large steel box during roastlng. It was cut into short temporary food service and regulatory measures in case small pleCeS and served at 10 o'clock in the momlng. Some of possible occurrences of food poISOnlng. noted that some portion of the meat was rare; i.e., it was insufficiently cooked. Laboratory and other epidemiologlCal data will be published ln the outbreak, a total 58 cases were counted. There were in Infectious Agents Surveillance Report, vol. 22 (June, 200 I ). 41 primary infections, 1 1 secondary infections including a We thank the clinical institutions, schools and other institu- case which took place in a nursery school, and 6 cases whose tions fわr their collaboration. Laboratory and Epidemiology Communications GB Virus C/Hepatitis G Virus Does Not Induce Expression of p44 Antigen in Chimpanzee Hepatocytes Yohko K. Shimizu*, Minako Hijikata and Hiroshi Yoshikural Department ofRespiratoyy Diseases, Research Institute, International Medical Center ofJapan, Toyama 1-21-1 Shinjuku-ku, Too,0 162-8655 and JNational Institute ofInfectious Diseases, Toyama 1-23-1, Shinjuku-ku, Tokyo 162-8640 Communicated by Hiroshi Ybshikura (Accepted June 1, 2001) The cytoplasmic antlgen, p44, was orlglnally discovered found chimpanzees whose sera were positive for GBV-C什IGV in hepatocytes of chimpanzees experimentally infected with RNA by RTIPCR. -
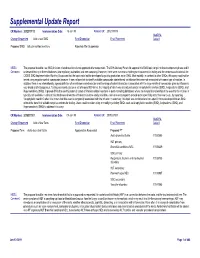
Detail Report
Supplemental Update Report CR Number: 2012319113 Implementation Date: 16-Jan-19 Related CR: 2012319113 MedDRA Change Requested Add a new SMQ Final Disposition Final Placement Code # Proposed SMQ Infusion related reactions Rejected After Suspension MSSO The proposal to add a new SMQ Infusion related reactions is not approved after suspension. The ICH Advisory Panel did approve this SMQ topic to go into the development phase and it Comment: underwent testing in three databases (two regulatory authorities and one company). However, there were numerous challenges encountered in testing and the consensus decision of the CIOMS SMQ Implementation Working Group was that the topic could not be developed to go into production as an SMQ. Most notably, in contrast to other SMQs, this query could not be tested using negative control compounds because it was not possible to identify suitable compounds administered via infusion that were not associated with some type of reaction. In addition, there is no internationally agreed definition of an infusion related reaction and the range of potential reactions associated with the large variety of compounds given by infusion is very broad and heterogenous. Testing was conducted on a set of around 500 terms, the majority of which was already included in Anaphylactic reaction (SMQ), Angioedema (SMQ), and Hypersensitivity (SMQ). It proved difficult to identify potential cases of infusion related reactions in post-marketing databases where the temporal relationship of the event to the infusion is typically not available. In clinical trial databases where this information is more easily available, users are encouraged to provide more specificity about the event, e.g., by reporting “Anaphylactic reaction” when it is known that this event is temporally associated with the infusion. -

Download Article PDF/Slides
Kan Lu, PharmD New Antiretrovirals for Based on a presentation at prn by Roy M. Gulick, md, mph the Treatment of HIV: Kan Lu, PharmD | Drug Development Fellow University of North Carolina School of Pharmacy Chapel Hill, North Carolina The View in 2006 Roy M. Gulick, md, mph Reprinted from The prn Notebook® | october 2006 | Dr. James F. Braun, Editor-in-Chief Director, Cornell Clinical Trials Unit | Associate Professor of Medicine, Meri D. Pozo, PhD, Managing Editor. Published in New York City by the Physicians’ Research Network, Inc.® Weill Medical College of Cornell University | New York, New York John Graham Brown, Executive Director. For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©october 2006 substantial progress continues to be made in the arena of cokinetics and a long extracellular half-life of approximately 10 hours antiretroviral drug development. prn is again proud to present its annual (Zhu, 2003). During apricitabine’s development, a serious drug interac- review of the experimental agents to watch for in the coming months and tion with lamivudine (Epivir) was noted. Although the plasma years. This year’s review is based on a lecture by Dr. Roy M. Gulick, a long- concentrations of apricitabine were unaffected by coadministration of time friend of prn, and no stranger to the antiretroviral development lamivudine, the intracellular concentrations of apricitabine were reduced pipeline. by approximately sixfold. Additionally, the 50% inhibitory concentration To date, twenty-two antiretrovirals have been approved by the Food (ic50) of apricitabine against hiv with the M184V mutation was increased and Drug Administration (fda) for the treatment of hiv infection. -

Cenicriviroc CSF Abstract Page 1 of 3 Title: Cerebrospinal Fluid Exposure
Cenicriviroc CSF Abstract Title: Cerebrospinal fluid exposure of cenicriviroc in HIV-positive individuals with cognitive impairment Authors: Jasmini Alagaratnam1, Laura Else2, Sujan Dilly Penchala2, Elizabeth Challenger2, Ken Legg1, Claire Petersen1, Brynmor Jones3, Ranjababu Kulasegaram4, Star Seyedkazemi5, Eric Lefebvre5, Saye Khoo2 and Alan Winston1 1. Division of Infectious Diseases, Department of Medicine, Imperial College London, London W2 1PG 2. Department of Pharmacology, University of Liverpool, Liverpool L69 7SX, UK 3. Department of Radiology, Imperial College Healthcare NHS Trust, London W2 1NY 4. St. Thomas’ Hospital, London W6 8RP, UK 5. Clinical Development, Allergan plc, South San Francisco, USA Page 1 of 3 Cenicriviroc CSF Abstract Abstract Background: Cenicriviroc, a dual C-C chemokine receptor type 2 (CCR2) and type 5 (CCR5) antagonist, is a potential adjunctive therapy, along with antiretroviral therapy (ART), for the management of HIV-associated cognitive disorders. Materials and Methods: Virologically suppressed persons living with HIV (PLWH) with a clinical diagnosis of HIV-related cognitive impairment intensified ART with cenicriviroc once daily, dose dependent on current ART, for eight weeks. Subjects with current or previous use of CCR5 inhibitors were not eligible. We assessed cerebrospinal fluid (CSF) exposure of cenicriviroc and CSF albumin at week 8, and changes in cognitive function over 8 weeks. Cenicriviroc concentration was determined using reverse phase high-performance liquid chromatography (HPLC) with geometric mean (GM) and 95% confidence intervals (CI) calculated. The proposed cenicriviroc target concentration was above the 90% effective concentration (EC90) for cenicriviroc (0.17 ng/mL), with the lower limit of quantification (LLQ) 0.24 ng/mL taken as target concentration. -

Coxiella Burnetii
SENTINEL LEVEL CLINICAL LABORATORY GUIDELINES FOR SUSPECTED AGENTS OF BIOTERRORISM AND EMERGING INFECTIOUS DISEASES Coxiella burnetii American Society for Microbiology (ASM) Revised March 2016 For latest revision, see web site below: https://www.asm.org/Articles/Policy/Laboratory-Response-Network-LRN-Sentinel-Level-C ASM Subject Matter Expert: David Welch, Ph.D. Medical Microbiology Consulting Dallas, TX [email protected] ASM Sentinel Laboratory Protocol Working Group APHL Advisory Committee Vickie Baselski, Ph.D. Barbara Robinson-Dunn, Ph.D. Patricia Blevins, MPH University of Tennessee at Department of Clinical San Antonio Metro Health Memphis Pathology District Laboratory Memphis, TN Beaumont Health System [email protected] [email protected] Royal Oak, MI BRobinson- Erin Bowles David Craft, Ph.D. [email protected] Wisconsin State Laboratory of Penn State Milton S. Hershey Hygiene Medical Center Michael A. Saubolle, Ph.D. [email protected] Hershey, PA Banner Health System [email protected] Phoenix, AZ Christopher Chadwick, MS [email protected] Association of Public Health Peter H. Gilligan, Ph.D. m Laboratories University of North Carolina [email protected] Hospitals/ Susan L. Shiflett Clinical Microbiology and Michigan Department of Mary DeMartino, BS, Immunology Labs Community Health MT(ASCP)SM Chapel Hill, NC Lansing, MI State Hygienic Laboratory at the [email protected] [email protected] University of Iowa [email protected] Larry Gray, Ph.D. Alice Weissfeld, Ph.D. TriHealth Laboratories and Microbiology Specialists Inc. Harvey Holmes, PhD University of Cincinnati College Houston, TX Centers for Disease Control and of Medicine [email protected] Prevention Cincinnati, OH om [email protected] [email protected] David Welch, Ph.D. -

Cytomegalovirus Infection of the Human Gastrointestinal Tract
Journal of Gastroenterology and Hepatology (1999) 14, 973–976 OESOPHAGOGASTRODUODENAL DISORDERS Cytomegalovirus infection of the human gastrointestinal tract SUSAMA PATRA, SUBASH C SAMAL, ASHOK CHACKO, VADAKENADAYIL I MATHAN1 AND MINNIE M MATHAN1 The Wellcome Trust Research Laboratory, Department of Gastrointestinal Sciences, Christian Medical College and Hospital,Vellore,India Abstract Background: Current interest in cytomegalovirus (CMV) is largely due to an increase in the number of cases of acquired immunodeficiency syndrome and organ transplantation in recent years.The proper recognition of CMV-infected cells in gastrointestinal mucosal biopsies is critical for effective treatment of this condition. Methods: A total of 6580 endoscopic mucosal biopsies from 6323 patients in the 8-year period (1989–1996) were examined for CMV inclusion bodies. The endoscopic appearance and particularly the presence of ulcers were also analysed. Results and Conclusions: The prevalence of cytomegalovirus (CMV) inclusions was 9 per thousand in the gastrointestinal mucosal biopsies from an unselected group of patients. Of the 54 patients with CMV infection, 37 were immunocompromised and 17 apparently immunocompetent. Typical Cowdry inclusions and atypical inclusions were present, the latter more frequently in immunocompromised patients. The maximum prevalence of inclusions was in the oesophageal mucosa in immunocompro- mised individuals. © 1999 Blackwell Science Asia Pty Ltd Key words: cytomegalovirus, gastrointestinal tract, immunocompetent, immunocompromised, inclu- sion bodies, mucosal biopsies. INTRODUCTION in haematoxylin and eosin (HE)-stained histological samples is regarded as being sensitive and specific for Cytomegalovirus (CMV), first described in 1956,1 is a CMV infection,6–9 especially for samples from the gas- double-stranded DNA virus belonging to the herpes trointestinal tract. -

Red, Weeping and Oozing P.51 6
DERM CASE Test your knowledge with multiple-choice cases This month–9 cases: 1. Red, Weeping and Oozing p.51 6. A Chronic Condition p.56 2. A Rough Forehead p.52 7. “Why am I losing hair?” p.57 3. A Flat Papule p.53 8. Bothersome Bites p.58 4. Itchy Arms p.54 9. Ring-like Rashes p.59 5. A Patchy Neck p.55 on © buti t ri , h ist oad rig D wnl Case 1 y al n do p ci ca use o er sers nal C m d u rso m rise r pe o utho y fo C d. A cop or bite ngle Red, Weleepirnohig a sind Oozing a se p rint r S ed u nd p o oris w a t f uth , vie o Una lay AN12-year-old boy dpriesspents with a generalized, itchy rash over his body. The rash has been present for two years. Initially, the lesions were red, weeping and oozing. In the past year, the lesions became thickened, dry and scaly. What is your diagnosis? a. Psoriasis b. Pityriasis rosea c. Seborrheic dermatitis d. Atopic dermatitis (eczema) Answer Atopic dermatitis (eczema) (answer d) is a chroni - cally relapsing dermatosis characterized by pruritus, later by a widespread, symmetrical eruption in erythema, vesiculation, papulation, oozing, crust - which the long axes of the rash extend along skin ing, scaling and, in chronic cases, lichenification. tension lines and give rise to a “Christmas tree” Associated findings can include xerosis, hyperlin - appearance. Seborrheic dermatitis is characterized earity of the palms, double skin creases under the by a greasy, scaly, non-itchy, erythematous rash, lower eyelids (Dennie-Morgan folds), keratosis which might be patchy and focal and might spread pilaris and pityriasis alba.