The Ribosome and Its Role in Protein Folding: Looking Through a Magnifying Glass
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hexosamine Biosynthetic Pathway-Derived O-Glcnacylation Is Critical for RANKL-Mediated Osteoclast Differentiation
International Journal of Molecular Sciences Article Hexosamine Biosynthetic Pathway-Derived O-GlcNAcylation Is Critical for RANKL-Mediated Osteoclast Differentiation Myoung Jun Kim 1,†, Hyuk Soon Kim 2,3,† , Sangyong Lee 1, Keun Young Min 1, Wahn Soo Choi 1,4 and Jueng Soo You 1,4,* 1 School of Medicine, Konkuk University, Seoul 05029, Korea; [email protected] (M.J.K.); [email protected] (S.L.); [email protected] (K.Y.M.); [email protected] (W.S.C.) 2 Department of Biomedical Sciences, College of Natural Science, Dong-A University, Busan 49315, Korea; [email protected] 3 Department of Health Sciences, The Graduate School of Dong-A University, Busan 49315, Korea 4 KU Open Innovation Center, Research Institute of Medical Science, Konkuk University, Chungju 27478, Korea * Correspondence: [email protected]; Tel.: +82-2-2049-6235 † The first two authors are equally contributed. Abstract: O-linked-N-acetylglucosaminylation (O-GlcNAcylation) performed by O-GlcNAc trans- ferase (OGT) is a nutrient-responsive post-translational modification (PTM) via the hexosamine biosynthetic pathway (HBP). Various transcription factors (TFs) are O-GlcNAcylated, affecting their activities and significantly contributing to cellular processes ranging from survival to cellular dif- ferentiation. Given the pleiotropic functions of O-GlcNAc modification, it has been studied in various fields; however, the role of O-GlcNAcylation during osteoclast differentiation remains to be explored. Kinetic transcriptome analysis during receptor activator of nuclear factor-kappaB (NF-κB) ligand (RANKL)-mediated osteoclast differentiation revealed that the nexus of major nutri- ent metabolism, HBP was critical for this process. We observed that the critical genes related to HBP Citation: Kim, M.J.; Kim, H.S.; activation, including Nagk, Gfpt1, and Ogt, were upregulated, while the global O-GlcNAcylation was Lee, S.; Min, K.Y.; Choi, W.S.; You, J.S. -

5 Inhibitors of Protein Synthesis
5 Inhibitors of protein synthesis Many antimicrobial substances inhibit protein biosynthesis. In most cases the inhibition involved one or other of the events which take place on the ribosomes. Only a few agents inhibit either amino acid activation or the attachment of the activated amino acid to the terminal adenylic acid residue of transfer RNA (tRNA). There are many chemical types to be found among the inhibitors of prolein synthesis, a fa ct which has increased the difficulty of unders tanding the molecular nature of their inhibitory effects. Indeed, while the reaction which is inhibited has been ideIHified with some precision in certain instances, the nature of the molecular interaction between the sensitive site and inhibi tor remains generally elusive. The reason lies in the complexity of the reactions leading to the for mation of correctly sequenced polypeptides on the ribosome and also in the complex. ity of the structure of the ribosome itself. Our intention is to provide an outline of the current knowledge of the steps in protein biosynthesis. More detailed discussion is given to those specific reactions which are blocked by the inhibitors of protein biosynthesis. RIBOSOMES These remarkable organelles are the machines upon which polypeptides are elaborated. There are three main classes of ribosomes identified by their sedimentation coefficients. The 80S ribosomes are apparently confined to eukaryotic cells, while 70S ribosomes are found in both prokaryotic and euk aryotic cells. A unique species of50-55S ribosome found only in mamma· tian mitochondria resembles bacterial ribosomes in functional organization and antibiotic sensitivity. The 80S particle dissociates reversibly into 60S and 405 subunits and the 70S into 505 and 305 subunits as the Mg:2+ concentration of the solution is reduced. -

Techniques for Study of Protein Synthesis
283. TECHNIQUES FOR STUDY OF PROTEIN SYNTHE'SIS F. C. PARRISH, JR. IOWA STATE UNIVERSITY ............................................................................... A study of skeletal muscle protein biosynthesis requires a number of both simple and sophisticated techniques because it involves a study of cellu- lar subunits and molecules and their reactions. Many of these techniques h?-ve already been successfully used to acquire a considerable body of knowledge about muscle biochemistry and ultrastructure. Consequently, adaptation of these techniques provides a strong potential for obtaining some very inter- esting and illuminating information about muscle protein biosynthesis and development. Other aids in the study of muscle protein biosynthesis hwe been the knowledge supplied by the molecular biologist on the biosynthetic mecha- nism of cells and on the behavior of actin and myosin in solution. Recent research, utilizing many of the same techniques as those used for the study of protein chemistry and structure, has provided us with some very profound information about myof ibrillogenesis of mammalian skeletal muscle. Naturally, the subject of myofibrillogenesis is of much interest and concern to the muscle biologist and meat scientist because the proteins of the myofibril are the ones that most directly affect muscle growth and development, contraction, rigor mortis, meat tenderness, water binding, emulsification and human nutri- tion. If we are able to make substantial improvement in the quantitative, qualitative, and nutritive characteristics of meat we must turn to the use of techniques that will yield information on how muscle proteins are synthesized and formed into meat at the cellular and molecular level. With knowledge gained at these levels we can then begin to regulate those mechanisms and compounds affecting muscle growth and composition. -

Translation and Folding of Single Proteins in Real Time PNAS PLUS
Translation and folding of single proteins in real time PNAS PLUS Florian Wrucka,1, Alexandros Katranidisb,2, Knud H. Nierhausc,3, Georg Büldtb,d, and Martin Hegnera,2 aCentre for Research on Adaptive Nanostructures and Nanodevices, School of Physics, Trinity College Dublin, Dublin 2, Ireland; bInstitute of Complex Systems ICS-5, Forschungszentrum Jülich, 52425 Jülich, Germany; cInstitute for Medical Physics and Biophysics, Charité–Universitätsmedizin Berlin, 10117 Berlin, Germany; and dLaboratory for Advanced Studies of Membrane Proteins, Moscow Institute of Physics and Technology, 141700 Dolgoprudny, Russia Edited by George H. Lorimer, University of Maryland, College Park, MD, and approved April 21, 2017 (received for review October 27, 2016) Protein biosynthesis is inherently coupled to cotranslational pro- ensemble methods. Optical tweezers have been used to observe tein folding. Folding of the nascent chain already occurs during stepping of motor proteins (19–23), DNA–protein complexes (24), synthesis and is mediated by spatial constraints imposed by the as well as unfolding and refolding of RNA molecules and proteins ribosomal exit tunnel as well as self-interactions. The polypeptide’s (25, 26). This powerful single-molecule method has provided in- vectorial emergence from the ribosomal tunnel establishes the possi- formation on (i) the translation machinery by reporting on the ble folding pathways leading to its native tertiary structure. How strength of interactions between the ribosome and mRNA (27), cotranslational protein folding and the rate of synthesis are linked (ii) its translocation along a short hairpin-forming mRNA mole- to a protein’s amino acid sequence is still not well defined. Here, we cule (28), as well as (iii) the release of an arrested nascent chain follow synthesis by individual ribosomes using dual-trap optical twee- (7). -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Review Article Intracellular Protein Biosynthesis
Review Article Intracellular Protein Biosynthesis: A Review Abstract Proteins are macromolecules made up of many amino acids that linked together by peptide bond to make a protein molecule. The sequence and the number of amino acids determines each protein unique structure and specific function. Proteins play a vital role in living systems and play important biological functions. Biosynthesis of protein occur in our body cells in order to support the biological function in our body. Intracellular protein synthesis is a complex process that involve the transformation of information and instructions from a genetic material DNA inside the nucleus to form mRNA molecules that transferred to the cytoplasm and liked to the cytoplasmic ribosome. Subsequently, the m RNA and further encode a sequence of amino acid in a specific order and number to form a polypeptide chains that finally undergoes conformational changes and folding to form a particular structure protein. This review will focus on the tow consecutive stages of protein biosynthesis; transcription and translation, and their substage processes; initiation, elongation, and termination. Briefly, overview the role of protein in the biological function and the different types of protein structure. Keywords: Proteins;Amino Acids; Peptide; Transcription; Translation. 1. INTRODUCTION Proteins are macromolecules that consist of one or more chains of amino acids that are linked together by peptide boundaries in a specific order. There are 20 different types of amino acids, and the order and number in which the different amino acids are arranged helps to determine the role of this particular protein. Proteins play a crucial role in the normal functioning of cells. -

Organization and Evolution of 5S Ribosomal Dna in the Fish Genome
In: Focus on Genome Research ISSN 1-59033-960-6 Editor: Clyde R. Williams, pp335-363 ©2004 Nova Science Publishers, Inc. Chapter X ORGANIZATION AND EVOLUTION OF 5S RIBOSOMAL DNA IN THE FISH GENOME Cesar Martins 1 and Adriane Pinto Wasko 2 Departamento de Morfologia, Instituto de Biociências, Universidade Estadual Paulista, CEP 18618-000, Botucatu, SP, Brazil. Phone/Fax +55 14 38116264, 1e-mail [email protected]; 2e-mail: [email protected] ABSTRACT In higher eukaryotes, the 5S ribosomal multigene family (5S rDNA) is tandemly organized in repeat units composed of a coding region (5S rRNA gene) and a non-transcribed spacer sequence (NTS). Although the 5S rDNA organization has been investigated in several vertebrate species, present data are concentrated in mammals and amphibians, whereas other groups, such as fishes, have been poorly studied. To further the understanding on the dynamics and evolution of 5S rDNA arrays in the vertebrate genome, recent studies have focused on the genome organization of these sequences in fish species, which represent the base group of vertebrate evolution. It was evidenced that the chromosome distribution of the 5S rDNA is quite conserved among related fish species occupying an interstitial position in the chromosomes. Although the 5S rDNA clusters have been maintained conserved in the chromosomes, changes in the nucleotide sequences and organization of the repeat units have occurred in fish species, as demonstrated by the presence of 5S rDNA variant types within and between genomes clustered in distinct chromosome environments. These variants are distributed in two major classes, suggesting that such pattern could represent a primitive condition for the fish genome, as well as for vertebrates. -
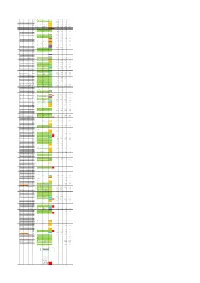
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

Trigger Factor in Complex with the Ribosome Forms a Molecular Cradle
letters to nature gel electrophoresis and NMR) and binds NC with the same affinity and stoichiometry 13. Kim, C.-H. & Tinoco, I. Jr. A retroviral RNA kissing complex containing only two G–C base pairs. observed for the native WCES RNA14. Proc. Natl Acad. Sci. USA 97, 9396–9401 (2000). 14. D’Souza, V. et al. Identification of a high-affinity nucleocapsid protein binding site within the Sample preparation Moloney murine leukemia virus W-RNA packaging signal. Implications for genome recognition. MoMuLV NC protein and RNA constructs were prepared as described14,15. RNAs of 35 J. Mol. Biol. 314, 217–232 (2001). nucleotides or less were obtained from Dharmacon and purified by denaturing gel 15. D’Souza, V., Dey, A., Habib, D. & Summers, M. F. NMR structure of the 101 nucleotide core electrophoresis. Samples for all NMR, ITC and polyacrylamide gel electrophoresis (PAGE) encapsidation signal of the Moloney murine leukemia virus. J. Mol. Biol. 337, 427–442 (2004). measurements were prepared in Tris-HCl buffer (10 mM at pH 7.0, 10 mM NaCl, 0.1 mM 16. De Guzman, R. N. et al. Structure of the HIV-1 nucleocapsid protein bound to the SL3 W-RNA recognition element. Science 279, 384–388 (1998). ZnCl2 and 0.1 mM b-mercaptoethanol). 17. Amarasinghe, G. K. et al. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 NC binding experiments of the W-RNA packaging signal. J. Mol. Biol. 301, 491–511 (2000). 18. Schuller, W., Dong, C.-Z., Wecker, K. & Roques, B.-P.NMR structure of the complex between the zinc ITC data (VP-ITC calorimeter, MicroCal Corp.) were measured at 30 8C. -

HUMAN RIBOSOME BIOGENESIS and the REGULATION of the TUMOUR SUPPRESSOR P53
HUMAN RIBOSOME BIOGENESIS AND THE REGULATION OF THE TUMOUR SUPPRESSOR p53 Andria Pelava Submitted for Doctor of Philosophy Final submission: December 2016 Institute of Cell and Molecular Biosciences Faculty of Medical Sciences Newcastle University ii Abstract Ribosome production is an energetically expensive and, therefore, highly regulated process. Defects in ribosome biogenesis lead to genetic diseases called Ribosomopathies, such as Dyskeratosis Congenita (DC), and mutations in ribosomal proteins and ribosome biogenesis factors are linked to multiple types of cancer. During ribosome biogenesis, the ribosomal RNAs (rRNAs) are processed and modified, and defects in ribosome biogenesis lead to the activation of p53. This project aimed to investigate the functions of Dyskerin, mutated in X-linked DC, in human ribosome biogenesis and p53 regulation, and to explore the link between ribosome production and p53 homeostasis. Dyskerin is an rRNA pseudouridine synthase and required for telomere maintenance. There is some debate as to whether DC is the result of telomere maintenance or ribosome biogenesis defects. It is shown here that human Dyskerin is required for the production of both LSU and SSU, and knockdown of Dyskerin leads to p53 activation via inhibition of MDM2 via the 5S RNP, an LSU assembly intermediate which accumulates after ribosome biogenesis defects. My data indicate that p53 activation, due to defects in ribosome biogenesis, may contribute to the clinical symptoms seen in patients suffering with DC. In addition, it is shown that defects in early or late stages of SSU or LSU biogenesis, result in activation of p53 via the 5S RNP-MDM2 pathway, and that p53 is induced in less than 12 hours after ribosome biogenesis defects. -

Selenoprotein H Is an Essential Regulator of Redox Homeostasis That
Selenoprotein H is an essential regulator of redox PNAS PLUS homeostasis that cooperates with p53 in development and tumorigenesis Andrew G. Coxa, Allison Tsomidesa, Andrew J. Kima, Diane Saundersa, Katie L. Hwanga, Kimberley J. Evasonb, Jerry Heidelc, Kristin K. Brownd, Min Yuand, Evan C. Liend, Byung Cheon Leea,e, Sahar Nissima, Bryan Dickinsonf, Sagar Chhangawalag, Christopher J. Changh,i, John M. Asarad, Yariv Houvrasg, Vadim N. Gladysheva,j, and Wolfram Goesslinga,j,k,l,1 aBrigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; bUniversity of Utah, Salt Lake City, UT 84112; cOregon State University, Corvallis, OR 97331; dBeth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115; eKorea University, 02841 Seoul, Republic of Korea; fUniversity of Chicago, Chicago, IL 60637; gWeill Cornell Medical College and New York Presbyterian Hospital, New York, NY 10065; hHoward Hughes Medical Institute, Bethesda, MD 20815; iUniversity of California, Berkeley, CA 20815; jBroad Institute of MIT and Harvard, Cambridge, MA 02142; kHarvard Stem Cell Institute, Cambridge, MA 02138; and lDana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02115 Edited by Leonard I. Zon, Howard Hughes Medical Institute, Boston Children’s Hospital, Harvard Medical School, Boston, MA, and accepted by Editorial Board Member Carol Prives July 18, 2016 (received for review January 7, 2016) Selenium, an essential micronutrient known for its cancer preven- mice exhibit impaired selenium transport from the liver to pe- tion properties, is incorporated into a class of selenocysteine- ripheral tissues and show growth retardation and impaired motor containing proteins (selenoproteins). Selenoprotein H (SepH) is a coordination (13, 14). -

PROTEIN SYNTHESIS and PROCESSING Protein Biosynthesis
PROTEIN SYNTHESIS AND PROCESSING Protein biosynthesis is the process whereby biological cells generate new proteins; it is balanced by the loss of cellular proteins via degradation or export. Translation, the assembly of amino acids by ribosomes, is an essential part of the biosynthetic pathway, along with generation of messenger RNA (mRNA), aminoacylation of transfer RNA (tRNA), co- translational transport, and post-translational modification. Protein biosynthesis is strictly regulated at multiple steps. They are principally during transcription (phenomena of RNA synthesis from DNA template) and translation (phenomena of amino acid assembly from RNA). The cistron DNA is transcribed into the first of a series of RNA intermediates. The last version is used as a template in synthesis of a polypeptide chain. Protein will often be synthesized directly from genes by translating mRNA. However, when a protein must be available on short notice or in large quantities, a protein precursor is produced. A proprotein is an inactive protein containing one or more inhibitory peptides that can be activated when the inhibitory sequence is removed byproteolysis during posttranslational modification. A preprotein is a form that contains a signal sequence (an N-terminal signal peptide) that specifies its insertion into or through membranes, i.e., targets them for secretion. The signal peptide is cleaved off in the endoplasmic reticulum.[1] Preproproteins have both sequences (inhibitory and signal) still present. In protein synthesis, a succession of tRNA molecules charged with appropriate amino acids are brought together with an mRNA molecule and matched up by base-pairing through the anti-codons of the tRNA with successive codons of the mRNA.