RELA Mutations Are Associated to Common Variable Immunodeficiency and Systemic Lupus Erythematosus Hicham Lamrini
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cytokine Nomenclature
RayBiotech, Inc. The protein array pioneer company Cytokine Nomenclature Cytokine Name Official Full Name Genbank Related Names Symbol 4-1BB TNFRSF Tumor necrosis factor NP_001552 CD137, ILA, 4-1BB ligand receptor 9 receptor superfamily .2. member 9 6Ckine CCL21 6-Cysteine Chemokine NM_002989 Small-inducible cytokine A21, Beta chemokine exodus-2, Secondary lymphoid-tissue chemokine, SLC, SCYA21 ACE ACE Angiotensin-converting NP_000780 CD143, DCP, DCP1 enzyme .1. NP_690043 .1. ACE-2 ACE2 Angiotensin-converting NP_068576 ACE-related carboxypeptidase, enzyme 2 .1 Angiotensin-converting enzyme homolog ACTH ACTH Adrenocorticotropic NP_000930 POMC, Pro-opiomelanocortin, hormone .1. Corticotropin-lipotropin, NPP, NP_001030 Melanotropin gamma, Gamma- 333.1 MSH, Potential peptide, Corticotropin, Melanotropin alpha, Alpha-MSH, Corticotropin-like intermediary peptide, CLIP, Lipotropin beta, Beta-LPH, Lipotropin gamma, Gamma-LPH, Melanotropin beta, Beta-MSH, Beta-endorphin, Met-enkephalin ACTHR ACTHR Adrenocorticotropic NP_000520 Melanocortin receptor 2, MC2-R hormone receptor .1 Activin A INHBA Activin A NM_002192 Activin beta-A chain, Erythroid differentiation protein, EDF, INHBA Activin B INHBB Activin B NM_002193 Inhibin beta B chain, Activin beta-B chain Activin C INHBC Activin C NM005538 Inhibin, beta C Activin RIA ACVR1 Activin receptor type-1 NM_001105 Activin receptor type I, ACTR-I, Serine/threonine-protein kinase receptor R1, SKR1, Activin receptor-like kinase 2, ALK-2, TGF-B superfamily receptor type I, TSR-I, ACVRLK2 Activin RIB ACVR1B -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Secretome Screening Reveals Immunomodulating Functions Of
www.nature.com/scientificreports OPEN Secretome screening reveals immunomodulating functions of IFNα‑7, PAP and GDF‑7 on regulatory T‑cells Mei Ding1*, Rajneesh Malhotra2, Tomas Ottosson2, Magnus Lundqvist3, Aman Mebrahtu3, Johan Brengdahl1, Ulf Gehrmann2, Elisabeth Bäck4, Douglas Ross‑Thriepland5, Ida Isaksson6, Björn Magnusson1, Kris F. Sachsenmeier7, Hanna Tegel3, Sophia Hober3, Mathias Uhlén3, Lorenz M. Mayr5, Rick Davies5, Johan Rockberg3 & Lovisa Holmberg Schiavone1* Regulatory T cells (Tregs) are the key cells regulating peripheral autoreactive T lymphocytes. Tregs exert their function by suppressing efector T cells. Tregs have been shown to play essential roles in the control of a variety of physiological and pathological immune responses. However, Tregs are unstable and can lose the expression of FOXP3 and suppressive functions as a consequence of outer stimuli. Available literature suggests that secreted proteins regulate Treg functional states, such as diferentiation, proliferation and suppressive function. Identifcation of secreted proteins that afect Treg cell function are highly interesting for both therapeutic and diagnostic purposes in either hyperactive or immunosuppressed populations. Here, we report a phenotypic screening of a human secretome library in human Treg cells utilising a high throughput fow cytometry technology. Screening a library of 575 secreted proteins allowed us to identify proteins stabilising or destabilising the Treg phenotype as suggested by changes in expression of Treg marker proteins FOXP3 and/or CTLA4. Four proteins including GDF‑7, IL‑10, PAP and IFNα‑7 were identifed as positive regulators that increased FOXP3 and/or CTLA4 expression. PAP is a phosphatase. A catalytic‑dead version of the protein did not induce an increase in FOXP3 expression. -

Affiliations
Supplementary material Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1 *Nils Landegren1,2, Donald Sharon3,4, Eva Freyhult2,5,6,, Åsa Hallgren1,2, Daniel Eriksson1,2, Per-Henrik Edqvist7, Sophie Bensing8, Jeanette Wahlberg9, Lawrence M. Nelson10, Jan Gustafsson11, Eystein S Husebye12, Mark S Anderson13, Michael Snyder3, Olle Kämpe1,2 Nils Landegren and Donald Sharon contributed equally to the work Affiliations 1Department of Medicine (Solna), Karolinska University Hospital, Karolinska Institutet, Sweden 2Science for Life Laboratory, Department of Medical Sciences, Uppsala University, Sweden 3Department of Genetics, Stanford University, California, USA 4Department of Molecular, Cellular, and Developmental Biology, Yale University, Connecticut, USA 1 5Department of Medical Sciences, Cancer Pharmacology and Computational Medicine, Uppsala University 6Bioinformatics Infrastructure for Life Sciences 7Department of Immunology, Genetics and Pathology, Uppsala University, Sweden and Science for Life Laboratory 8 Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden 9Department of Endocrinology and Department of Medical and Health Sciences and Department of Clinical and Experimental Medicine, Linköping University, Linköping, Sweden 10Integrative Reproductive Medicine Group, Intramural Research Program on Reproductive and Adult Endocrinology, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA. 11Department -

Proteome-Wide Survey of the Autoimmune Target Repertoire In
www.nature.com/scientificreports OPEN Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine Received: 28 October 2015 Accepted: 23 December 2015 syndrome type 1 Published: 01 February 2016 Nils Landegren1,2,*, Donald Sharon3,4,*, Eva Freyhult2,5,6, Åsa Hallgren1,2, Daniel Eriksson1,2, Per-Henrik Edqvist7, Sophie Bensing8, Jeanette Wahlberg9, Lawrence M. Nelson10, Jan Gustafsson11, Eystein S Husebye12, Mark S Anderson13, Michael Snyder3 & Olle Kämpe1,2 Autoimmune polyendocrine syndrome type 1 (APS1) is a monogenic disorder that features multiple autoimmune disease manifestations. It is caused by mutations in the Autoimmune regulator (AIRE) gene, which promote thymic display of thousands of peripheral tissue antigens in a process critical for establishing central immune tolerance. We here used proteome arrays to perform a comprehensive study of autoimmune targets in APS1. Interrogation of established autoantigens revealed highly reliable detection of autoantibodies, and by exploring the full panel of more than 9000 proteins we further identified MAGEB2 and PDILT as novel major autoantigens in APS1. Our proteome-wide assessment revealed a marked enrichment for tissue-specific immune targets, mirroringAIRE ’s selectiveness for this category of genes. Our findings also suggest that only a very limited portion of the proteome becomes targeted by the immune system in APS1, which contrasts the broad defect of thymic presentation associated with AIRE-deficiency and raises novel questions what other factors are needed for break of tolerance. Autoimmune responses can ultimately be defined at the molecular level by the specific interaction between T- or B-cell receptors and a distinct self-molecule. In tissue-specific autoimmune disorders the immune system typically target molecules that are exclusively expressed in the affected tissue and involve a combined cellular and humoral response with cognate specificities1–3. -

Heatmaps - the Gene Expression Edition
Heatmaps - the gene expression edition Jeff Oliver 20 July, 2020 An application of heatmap visualization to investigate differential gene expression. Learning objectives 1. Manipulate data into a ‘tidy’ format 2. Visualize data in a heatmap 3. Become familiar with ggplot syntax for customizing plots Heatmaps for differential gene expression Heatmaps are a great way of displaying three-dimensional data in only two dimensions. But how can we easily translate tabular data into a format for heatmap plotting? By taking advantage of “data munging” and graphics packages, heatmaps are relatively easy to produce in R. Getting started We are going to start by isolating different types of information by imposing structure in our file managment. That is, we are going to put our input data in one folder and any output such as plots or analytical results in a different folder. We can use the dir.create to create these two folders: dir.create("data") dir.create("output") For this lesson, we will use a subset of data on a study of gene expression in cells infected with the influenza virus (doi: 10.4049/jimmunol.1501557). The study infected human plasmacytoid dendritic cells with the influenza virus, and compared gene expression in those cells to gene expression in uninfected cells. Thegoal was to see how the flu virus affected the function of these immune system cells. The data for this lesson is available at: http://tinyurl.com/flu-expression-data or https://jcoliver.github.io/ learn-r/data/GSE68849-expression.csv. Download this comma separated file and put it in the data folder. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Microarray Analysis of Colorectal Cancer Stromal Tissue Reveals Upregulation of Two Oncogenic Mirna Clusters
Published OnlineFirst March 27, 2012; DOI: 10.1158/1078-0432.CCR-11-1078 Clinical Cancer Human Cancer Biology Research Microarray Analysis of Colorectal Cancer Stromal Tissue Reveals Upregulation of Two Oncogenic miRNA Clusters Naohiro Nishida1,4, Makoto Nagahara2, Tetsuya Sato3, Koshi Mimori1, Tomoya Sudo1, Fumiaki Tanaka1, Kohei Shibata1, Hideshi Ishii1,4, Kenichi Sugihara2, Yuichiro Doki4, and Masaki Mori1,4 Abstract Purpose: Cancer stroma plays an important role in the progression of cancer. Although alterations in miRNA expression have been explored in various kinds of cancers, the expression of miRNAs in cancer stroma has not been explored in detail. Experimental Design: Using a laser microdissection technique, we collected RNA samples specific for epithelium or stroma from 13 colorectal cancer tissues and four normal tissues, and miRNA microarray and gene expression microarray were carried out. The expression status of miRNAs was confirmed by reverse transcriptase PCR. Furthermore, we investigated whether miRNA expression status in stromal tissue could influence the clinicopathologic factors. Results: Oncogenic miRNAs, including two miRNA clusters, miR-17-92a and miR-106b-25 cluster, were upregulated in cancer stromal tissues compared with normal stroma. Gene expression profiles from cDNA microarray analyses of the same stromal tissue samples revealed that putative targets of these miRNA clusters, predicted by Target Scan, such as TGFBR2, SMAD2, and BMP family genes, were significantly downregulated in cancer stromal tissue. Downregulated putative targets were also found to be involved in cytokine interaction and cellular adhesion. Importantly, expression of miR-25 and miR-92a in stromal tissues was associated with a variety of clinicopathologic factors. Conclusions: Oncogenic miRNAs were highly expressed in cancer stroma. -

Genomic Analyses of PMBL Reveal New Drivers and Mechanisms of Sensitivity to PD-1 Blockade
Regular Article LYMPHOID NEOPLASIA Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade Bjoern Chapuy,1,2,* Chip Stewart,3,* Andrew J. Dunford,3,* Jaegil Kim,3 Kirsty Wienand,1 Atanas Kamburov,3 Gabriel K. Griffin,4 Pei-Hsuan Chen,4 Ana Lako,4 Robert A. Redd,5 Claire M. Cote,1 Matthew D. Ducar,6 Aaron R. Thorner,6 Scott J. Rodig,4 Gad Getz,3,7,8,† Downloaded from https://ashpublications.org/blood/article-pdf/134/26/2369/1549702/bloodbld2019002067.pdf by Margaret Shipp on 30 December 2019 and Margaret A. Shipp1,† 1Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA; 2Department of Hematology and Oncology, University Medical Center Gottingen,¨ Gottingen,¨ Germany; 3Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, MA; 4Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA; 5Department of Data Sciences and 6Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, MA; and 7Department of Pathology and 8Center for Cancer Research, Massachusetts General Hospital, Harvard Medical School, Boston, MA KEY POINTS Primary mediastinal large B-cell lymphomas (PMBLs) are aggressive tumors that typically present as large mediastinal masses in young women. PMBLs share clinical, transcriptional, l Comprehensive genomic analyses of and molecular features with classical Hodgkin lymphoma (cHL), including constitutive PMBL reveal new activation of nuclear factor kB (NF-kB), JAK/STAT signaling, and programmed cell death genetic drivers such protein 1 (PD-1)–mediated immune evasion. The demonstrated efficacy of PD-1 blockade in as ZNF217. relapsed/refractory PMBLs led to recent approval by the US Food and Drug Administration l High mutational and underscored the importance of characterizing targetable genetic vulnerabilities in this burden, MSI, and disease. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -
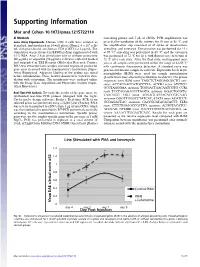
Supporting Information
Supporting Information Mor and Cohen 10.1073/pnas.1215722110 SI Methods containing primer and 5 μL of cDNA. PCR amplification was Gene Array Experiments. Human CD4 T cells were isolated as preceded by incubation of the mixture for 10 min at 95 °C, and described, and incubated in 24-well plates (Nunc), 4 × 106 cells/ the amplification step consisted of 45 cycles of denaturation, mL with plate-bound anti-human CD3 (OKT3) at 2 μg/mL. The annealing, and extension. Denaturation was performed for 15 s stimulation was performed in RPMI medium supplemented with at 95 °C; annealing was performed in 60 °C; and the extension 0.1% BSA. After 2 h of stimulation with or without cefuroxime was performed at 72 °C for 20 s, with fluorescence detection at (50 μg/mL) or ampicillin (50 μg/mL), cells were collected washed 72 °C after each cycle. After the final cycle, melting-point anal- and suspended in TRI Reagent (Molecular Research Center). yses of all samples were performed within the range of 62–95 °C RNA was extracted from samples and used to prepare probes for with continuous fluorescence detection. A standard curve was gene array in accord with the manufacturer’s instructions (Super- generated from one sample in each run. Expression levels of β2- Array Bioscience). Adequate labeling of the probes was tested microglobulin (B2M) were used for sample normalization before hybridization. Three healthy donors were tested in stim- (β-actin levels were affected by cefuroxime treatment). The primer ulation with cefuroxime. The membranes were analyzed online sequences were B2M sense TAGCTCTAGGAGGGCTG anti- with the Image Data Acquisition and Expression Analysis (Super- sense ACCACAACCATGCCTTA; ACVR2 sense ATCTCC- Array Bioscience).