Transmissible Spongiform Encephalopathies in Sheep: Experimental Determination of Genetic Susceptibility and Interspecies Transmission
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

License Contacts
License Contacts License Code Responsible License Status Date Applied Date Granted Date Expires Commodities Map References Area Parties Contact Details Office Type Communication Party Postal Address Physical Address Telephone Email Address 51522 14/2/2/1/2/ MC Active 01 April 2017 31 March 2019 BRM Namibia,J, K, 16.8153 Ha Onganja Mining Company Khomas,Windhoek; (Pty) Ltd (100%) Otjozondjupa,Okahan dja 51523 14/2/2/1/2/ MC Active 01 April 2017 31 March 2019 BRM Namibia,J, K, 18.1577 Ha Onganja Mining Company Khomas,Windhoek; (Pty) Ltd (100%) Otjozondjupa,Okahan dja 52604 14/2/2/1/2/ MC Pending 01 April 1994 31 March 2013 DS Picture Stone (Pty) Ltd Renewal (100%) 52605 14/2/2/1/2/ MC Pending 01 April 1994 31 March 2013 DS Picture Stone (Pty) Ltd Picture Stone (Pty) Ltd 0027-11-477- Renewal (100%) 6296 53182 14/2/2/1/2/ MC Pending 01 April 1974 31 March 2017 SPS Namibia,Hardap,Malta 7.3312 Ha Daniel Matheus Laufs Renewal hohe; P Truter (100%) 53979 14/2/2/1/2/ MC Active 01 April 2017 31 March 2019 BRM Namibia,K, 17.3954 Ha Robert Guy Carr (100%) Khomas,Windhoek 55669 14/2/2/1/2/ MC Pending 22 March 1983 31 March 2015 IM Namibia Mineral Renewal Development Company (Pty) Ltd (100%) 55926 14/2/2/1/2/ MC Pending 22 March 1983 31 March 2015 IM Namibia Mineral Renewal Development Company (Pty) Ltd (100%) 55927 14/2/2/1/2/ MC Pending 22 March 1983 31 March 2015 IM Namibia Mineral Namibia Mineral PO Box 24046, 28 Heinitzburg Street, 237055 [email protected] Renewal Development Company Development Company Windhoek, Khomas, Windhoek, Khomas, m.na (Pty) -

January/February 1996
Your high school library can have a free subscription to ANIMAL PEOPLE–– Nonprofit the only independent newspaper covering all the news about animal protection. Organization Send your acceptance to: U.S. Postage ANIMAL PEOPLE, POB 205, Shushan, NY 12873, or fax it to 518-854-9601. Paid ANIMAL PEOPLE has no alignment or affiliation with any advocacy organization. ANIMAL PEOPLE, Out of cod, Canada tells fishers "kill seals" Inc. ST. JOHNS, Newfoundland––Blaming harp seals for a 99% decline in the mass of spawning cod off the Atlantic coast of POB 205, SHUSHAN, NY 12873 Newfoundland, Canadian Fisheries Minister Brian Tobin on [ADDRESS CORRECTION REQUESTED.] December 18 moved to appease out-of-work cod fishers in his home province by expanding the 1996 seal killing quota to 250,000––actually higher than many annual quotas during the peak years of the seal hunt in the 1970s and early 1980s. In effect resuming the all-out seal massacres that prompt- ed international protest until clubbing newborn whitecoats and hunting seals from large vessels was suspended in 1983, Tobin also pledged to maintain a bounty of about 15¢ U.S. per pound for each dead seal landed, and said he would encourage the revived use of large vessels to help sealers attack seal breeding colonies on offshore ice floes. rassed by an International Fund for Animal Welfare campaign The prohibition on killing whitecoats remains in effect, worldwide to expose the lack of market demand for seal products. but only means young seals will be killed not as newborns but as A report on seal marketing strategy commissioned by the Canadian two-week-old beaters, just beginning to molt and crawl. -

Omdel Township 2020-2024 General Valuation Roll Erf No
OMDEL TOWNSHIP 2020-2024 GENERAL VALUATION ROLL ERF NO. SIZE M2 REGISTERED OWNER TITLE DEED NO. LAND VALUE N$ IMPROVEMENT VALUE N$ TOTAL VALUE N$ IMPROVEMENT DESCRIPTION ZONING STREET NAME TOWNSHIP EXTENSION 1 501 PHILLEMON J T2471/1990 27,555.00 64,700 92,255 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 2 513 HAUFIKU J T3553/2007 28,215.00 528,400 556,615 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 3 416 MARAKIA H T1879/1998 22,880.00 199,430 222,310 Dwelling & Bar SINGLE RESIDENTIAL MOSES GAROEB STREET OMDEL PROPER 4 416 HARAEB A T3125/2000 22,880.00 332,640 355,520 Dwelling SINGLE RESIDENTIAL MOSES GAROEB STREET OMDEL PROPER 5 572 NATIONAL HOUSING ENTERPRISES T976/1989 31,460.00 207,900 239,360 Dwelling SINGLE RESIDENTIAL MOSES GAROEB STREET OMDEL PROPER 6 721 KAMATI P T2313/1990 39,655.00 247,940 287,595 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 7 634 ZAARUS PENDA IMMANUEL T306/2017 34,870.00 238,050 272,920 Dwelling & Garage SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 8 679 DE KLERK E T2988/2010 37,345.00 350,350 387,695 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 9 943 LUCIANO IGNATIUS T2989/2010 51,865.00 97,000 148,865 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 10 589 SILAS NICODEMUS T2470/1990 32,395.00 70,100 102,495 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 11 693 JOSEF GEINGOB T5313/2014 38,115.00 97,000 135,115 Dwelling SINGLE RESIDENTIAL FESTUS NAHOLO STREET OMDEL PROPER 12 533 LUCAS J &Y E BOCK T2990/2010 -

Conference Program July 26-29, 2021 | Pacific Daylight Time 2021 Asee Virtual Conference President’S Welcome
CONFERENCE PROGRAM JULY 26-29, 2021 | PACIFIC DAYLIGHT TIME 2021 ASEE VIRTUAL CONFERENCE PRESIDENT’S WELCOME SMALL SCREEN, SAME BOLD IDEAS It is my honor, as ASEE President, to welcome you to the 128th ASEE Annual Conference. This will be our second and, almost certainly, final virtual conference. While we know there are limits to a virtual platform, by now we’ve learned to navigate online events to make the most of our experience. Last year’s ASEE Annual Conference was a success by almost any measure, and all of us—ASEE staff, leaders, volunteers, and you, our attendees—contributed to a great meeting. We are confident that this year’s event will be even better. Whether attending in person or on a computer, one thing remains the same, and that’s the tremendous amount of great content that ASEE’s Annual Conference unfailingly delivers. From our fantastic plenary speakers, paper presentations, and technical sessions to our inspiring lineup of Distinguished Lectures and panel discussions, you will have many learning opportunities and take-aways. I hope you enjoy this week’s events and please feel free to “find” me and reach out with any questions or comments! Sincerely, SHERYL SORBY ASEE President 2020-2021 2 Schedule subject to change. Please go to https://2021asee.pathable.co/ for up-to-date information. 2021 ASEE VIRTUAL CONFERENCE TABLE OF CONTENTS 2021 ASEE VIRTUAL CONFERENCE AND EXPOSITION PROGRAM ASEE BOARD OF DIRECTORS ................................................................................4 CONFERENCE-AT-A-GLANCE ................................................................................6 -

Human Origin Sites and the World Heritage Convention in Eurasia
World Heritage papers41 HEADWORLD HERITAGES 4 Human Origin Sites and the World Heritage Convention in Eurasia VOLUME I In support of UNESCO’s 70th Anniversary Celebrations United Nations [ Cultural Organization Human Origin Sites and the World Heritage Convention in Eurasia Nuria Sanz, Editor General Coordinator of HEADS Programme on Human Evolution HEADS 4 VOLUME I Published in 2015 by the United Nations Educational, Scientific and Cultural Organization, 7, place de Fontenoy, 75352 Paris 07 SP, France and the UNESCO Office in Mexico, Presidente Masaryk 526, Polanco, Miguel Hidalgo, 11550 Ciudad de Mexico, D.F., Mexico. © UNESCO 2015 ISBN 978-92-3-100107-9 This publication is available in Open Access under the Attribution-ShareAlike 3.0 IGO (CC-BY-SA 3.0 IGO) license (http://creativecommons.org/licenses/by-sa/3.0/igo/). By using the content of this publication, the users accept to be bound by the terms of use of the UNESCO Open Access Repository (http://www.unesco.org/open-access/terms-use-ccbysa-en). The designations employed and the presentation of material throughout this publication do not imply the expression of any opinion whatsoever on the part of UNESCO concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. The ideas and opinions expressed in this publication are those of the authors; they are not necessarily those of UNESCO and do not commit the Organization. Cover Photos: Top: Hohle Fels excavation. © Harry Vetter bottom (from left to right): Petroglyphs from Sikachi-Alyan rock art site. -
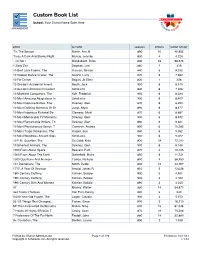
Custom Book List
Custom Book List School: Your District Name Goes Here MANAGEMENT BOOK AUTHOR LEXILE® POINTS WORD COUNT 'Tis The Season Martin, Ann M. 890 10 40,955 'Twas A Dark And Stormy Night Murray, Jennifer 830 4 4,224 ...Or Not? Mandabach, Brian 840 23 98,676 1 Zany Zoo Degman, Lori 860 1 415 10 Best Love Poems, The Hanson, Sharon 840 6 8,332 10 Coolest Dance Crazes, The Swartz, Larry 870 6 7,660 10 For Dinner Bogart, Jo Ellen 820 1 328 10 Greatest Accidental Inventi Booth, Jack 900 6 8,449 10 Greatest American President Scholastic 840 6 7,306 10 Mightiest Conquerors, The Koh, Frederick 900 6 8,034 10 Most Amazing Adaptations In Scholastic 900 6 8,409 10 Most Decisive Battles, The Downey, Glen 870 6 8,293 10 Most Defining Moments Of Th Junyk, Myra 890 6 8,477 10 Most Ingenious Fictional De Clemens, Micki 870 6 8,687 10 Most Memorable TV Moments, Downey, Glen 900 6 8,912 10 Most Remarkable Writers, Th Downey, Glen 860 6 9,321 10 Most Revolutionary Songs, T Cameron, Andrea 890 6 10,282 10 Most Tragic Romances, The Harper, Sue 860 6 9,052 10 Most Wondrous Ancient Sites Scholastic 900 6 9,022 10 P.M. Question, The De Goldi, Kate 830 18 72,103 10 Smartest Animals, The Downey, Glen 900 6 8,148 1000 Facts About Space Beasant, Pam 870 4 10,145 1000 Facts About The Earth Butterfield, Moira 850 6 11,721 1000 Questions And Answers Tames, Richard 890 9 38,950 101 Dalmatians, The Smith, Dodie 830 12 44,767 1777: A Year Of Decision Arnold, James R. -

Foodshare AR05
THE Heart of GREATER HARTFORD’S ANTI-HUNGER NETWORK 2005 ANNUAL REPORT From Our President Tragedy has a strange yet comforting way of mak- companies, foundations, civic groups and faith ing the world a smaller place. We are reminded that organizations whose generous contributions made it this is a world where we may suddenly need to turn possible for us to successfully close the door on our to our neighbors for help… and where we can help capital campaign, well, I just can’t thank you our neighbors in turn. enough. Hurricanes Katrina, Rita, and Wilma wreaked dev- Those aren’t the only doors I’m talking about, astation on a level rarely seen in our blessed country. however. For example: The immediate result was an inspiring response of •We’ve welcomed many new partner agencies to support and service from the public. Countless peo- the Foodshare network in recent years. The ple across the USA and beyond poured into the trend has been an average of 22 local anti- southern Gulf states to help those whose homes had hunger agencies a year added since 2002. been destroyed. Thousands in Connecticut freely gave to aid those in need, and Foodshare took part •Our third Food Industry Convoy of Caring for in the massive local effort. National Hunger Awareness Day last June was the biggest and best ever! More than 30 companies Hundreds and sometimes thousands of miles away, teamed up to make the 15-truck-long convoy a people opened their doors to those who, suddenly huge success. It may surprise you to know that homeless and jobless, began looking to other parts of the food industry now donates about 80% of the our country to start new lives. -

Buckaroo Book Award - by Author’S Name 1998 - 2020 Nominees
Buckaroo Book Award - by Author’s Name 1998 - 2020 Nominees Ada, Alma Flor - With Love, Little Red Hen [Nominee 2003] Agee, Jon - Life on Mars [Nominee 2020] Agee, Jon - My Rhinoceros [Nominee 2013] Allen, Elanna - Poor Little Guy [Nominee 2020] Andreae, Giles - Giraffes Can't Dance [Nominee 2002] Angleberger, Tom - McToad Mows Tiny Island [Nominee 2017] Applegate, Katherine - Ivan: The Remarkable True Story of the Shopping Mall Gorilla [Nominee 2016] Archambault, John & Martin, Bill Jr. - Chicka Chicka Boom Boom [Nominee 1999] Arnold, Tedd - Parts [1st Runner-Up 2003] Arnosky, Jim - Rattlesnake Dance [2nd Runner-Up 2003] Asher, Sandy - Too Many Frogs! [Nominee 2008] Auch, Mary Jane - Chickerella [Nominee 2008] Auch, Mary Jane - Easter Egg Farm [Nominee 2001] Bacon, Arthur and Henry - I Hate Reading: How to Get Through 20 Minutes of Reading a Day Without Really Reading [Nominee 2011] Bang, Molly - When Sophie Gets Angry [Nominee 2002] Banks, Kate - Max's Words [Nominee 2009] Bardhan-Quallen, Sudipta - Quackenstein Hatches a Family [Nominee 2012] Barnett, Mac - Extra Yarn [Nominee 2013] Barnett, Mac - President Taft is Stuck in the Bath [1st Runner-Up 2016] Barnett, Mac - Wolf, the Duck, and the Mouse [Nominee 2019] Barton, Chris - Shark vs. Train [1st Runner-Up 2012] Base, Graeme - Jungle Drums [Nominee 2006] Beaton, Kate - Princess and the Pony [2nd Runner-Up 2019] Beaumont, Karen - I Ain't Gonna Paint No More! [2nd Runner-up 2007] Beaumont, Karen - Move Over, Rover [Nominee 2009] Becker, Aaron - Journey [Nominee 2015] Becker, Bonny -

Arts Group New Home Work of Art Puppets, Persian Poetry, and Change
SUMMER HOURS: An The News will close INDEPENDENT Fridays at 1 p.m. JOURNAL of NEWS throughout the summer and OPINION until after Labor Day. YELLOW SPRINGS NEWS SINCE 1880 YELLOW SPRINGS, OHIO THURSDAY, JULY 8, 2010 VOLUME 131, NUMBER 27 PRICE: $1.25 Green towns offer new ideas By Lauren Heaton This is the second in a two-part report on As sustainability gains ground as an municipal energy conservation. integral component of city planning, many municipalities across the country are creat- restaurants, according to an article in Sus- ing ways to use less energy and ensure that tainable Industries magazine in March. the energy they use comes from renewable Oberlin, population 8,200, is one of 17 sources. cities around the world to join the nation’s Several cities researched for this article Climate Positive Development Program, have mandated that all new construction which will offer international partnership meets the Leadership in Energy and Envi- and technical support for the project. And as ronmental Design (LEED) energy-efficient an integral partner in the project, the city of standard. Other cities have established Oberlin has a $60,000 sustainability reserve funding mechanisms to provide incentives fund, generated through renewable energy for residential and commercial retrofits. credits purchased by Oberlin College, City The city of Oberlin, Ohio, and Oberlin Col- Manager Eric Norenberg said this week. lege have embarked on a green approach The city disperses the funds for efficiency to urban revitalization, while one tiny town projects, including seed money for a home in Kansas aims to become the country’s top weatherization program for low- to moder- model green community. -

Code 3 on Officer of the Month - October 2010 This Very Controversial Issue
CODE THREE A Palm Beach County P.B.A. Official Publication VOLUME 27 NUMBER 1 PUBLISHED QUARTERLY FOR MEMBERS February 2011 Governor Rick Scott’s pension proposal doesn’t match his prior statements during his campaign. http://www.youtube.com/watch?v=PudKNxTKzrc THE VOICE OF PALM BEACH COUNTY LAW ENFORCEMENT OFFICERS money to off-set their deficit on the “backs of the employees.” Please stay tuned for further comprehensive updates on all of President’s the above. In addition, I want to send our deepest, heartfelt condo- lences to the families of the two slain Miami-Dade officers, Message Amanda Haworth and Roger Castillo along with our fallen St. Petersburg brothers Sgt. Thomas Baitinger and Officer Jeffrey Yaslowitz, and also State Correctional Colonel Greg Malloy in John Kazanjian Holmes County. Our prayers are with all of their families at this time of mourning. Stay Safe. Be active. would like to wish everyone a healthy and a happy New Year. I We have recently completed our PBA Representative elec- tions and we have officially tallied the results. On behalf of the Executive Board, I’d like to personally thank the previous Representatives for all their dedicated services and welcome aboard all of our New Representatives. We’re grateful for all of their hard work. By way of update, we just got back from Tallahassee where the PBA was asked to make a presentation to a Senate Officer of the Month - September 2010 Committee overseeing changes in pensions. We were informed PBSO Cpl. John Sylvester by the Committee Chair, Senator Ring, that there will be a Nominated by: Sgt. -

Saint Louis Zoo Endowed Positions and Funds: a Growing Tradition
Saint Louis Zoo Endowed Positions and Funds: A Growing Tradition 1 About this book This book is about our wonderful Zoo donors who have established endowed funds and positions at the Saint Louis Zoo. Most importantly, it tells their stories. Stories about people who worked hard, built successful businesses in St. Louis, and wanted to give something back to their community. Stories about people who lived simply and quietly, and gave generous and unexpected gifts. People who made our city and our Zoo what it is today and whose vision will live on tomorrow. As author Alexander McCall Smith wrote, “A life without stories would be no life at all. And stories bound us, did they not, one to another, the living to the dead, people to animals, people to the land.” I am inspired by each of the stories in this book. Some are about people and names who might be familiar, some are less known. But I am struck by how each of these donors was touched by the Zoo at some point in his or her life and how each was inspired to make an investment in our future. As an anthropologist, I have studied landmark learning experiences, those “aha” moments when a spark is ignited and we are inspired to learn more, or a light bulb goes on, and we get it. We have seen something amazing, and we want to know more. For many, a visit to the Saint Louis Zoo is the first encounter with a wild animal. It is something memorable and wonderful, and that has not changed in more than 100 years. -

2019-2020 Annual Report (Pdf, 1
REPORT TO THE COMMUNITY July 2019-June 2020 Dear friends, 2020 proved to be one of the most unique and challenging years in the Science Museum of Minnesota’s long history as a trusted resource for the community’s science learning. Back in 1907, our founders set out to “promote among all classes of people the knowledge and enlightenment that are essential to right living and good citizenship.” Among the offerings that first year were lectures on the spread of contagious diseases, among many other topics. It’s fascinating to consider our organization’s roots in light of today’s events and the renewed interest in the science of public health. Looking back on our history, we are struck by what has changed over more than a century — and what remains the same. And perhaps most of all, we’re struck by the Science Museum’s impressive track record of evolving to meet the changing needs of the community. The museum continues to evolve. It’s part of who we are, and the challenges of the past year demanded it. We were on track to meet or even exceed our goals for FY20 when the pandemic hit. Like so many other businesses and organizations, we saw major revenue losses as a result of an extended closure to help slow the spread of COVID-19. We had to make extraordinarily difficult decisions to weather the crisis — decisions that resulted in a smaller organization and a significant reduction in staff. Despite challenges like we’ve never experienced before, the Science Museum remains deeply committed to serving our community through science and education that center equity.