Karyonim® Prenatal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TARGETED GENE PANELS and REFERENCE SEQUENCE (Refseq) TRANSCRIPTS by PHENOTYPE
ROYAL DEVON & EXETER NHS FOUNDATION TRUST Department of Molecular Genetics TARGETED GENE PANELS AND REFERENCE SEQUENCE (RefSeq) TRANSCRIPTS BY PHENOTYPE Table of Contents (Click to select) Alagille Syndrome 2 Chondrodysplasia punctata 2 Combined Pituitary Hormone Deficiency 2 Congenital Generalised Lipodystrophy 2 Congenital Hypothyroidism 2 Early-onset Diabetes and Autoimmunity 3 Endocrine Neoplasia Syndromes 3 Familial Glucocorticoid Deficiency 3 Familial Hyperparathyroidism 3 Familial Hypocalciuric Hypercalcaemia 3 Familial Hyperparathyroidism/hypercalcaemia 4 Familial Hypoparathyroidism 4 Familial Partial Lipodystrophy 4 Familial Porencephaly and HANAC syndrome 4 Familial Tumoral Calcinosis 4 Feingold syndrome 4 Gastrointestinal atresia 5 Generalised Arterial Calcification in Infancy 5 Holoprosencephaly 5 Hyperinsulinism 5 Hypophosphatemic Rickets 6 Isolated Growth Hormone Deficiency 6 Kabuki syndrome 6 Kallmann syndrome 6 Mandibulofacial Dysostosis with Microcephaly 6 Moebius syndrome 6 Monogenic Diabetes of the Young (MODY) 7 Multiple Exostosis 7 Neonatal Diabetes 8 Phaeochromocytoma/Paraganglioma 9 Pontocerebellar Hypoplasia 9 Primary pigmented nodular adrenocortical disease 9 Pseudohypoaldosteronism 9 Spondylocostal Dysostosis 9 Visceral heterotaxy 10 Page 1 of 10 ROYAL DEVON & EXETER NHS FOUNDATION TRUST Department of Molecular Genetics Alagille Syndrome Transcript(s) JAG1 NM_000214 NOTCH2 NM_024408 Chondrodysplasia punctata Transcript(s) AGPS NM_003659 ARSE NM_000047 EBP NM_006579 GNPAT NM_014236 PEX7 NM_000288 Combined Pituitary -

WES Gene Package Multiple Congenital Anomalie.Xlsx
Whole Exome Sequencing Gene package Multiple congenital anomalie, version 5, 1‐2‐2018 Technical information DNA was enriched using Agilent SureSelect Clinical Research Exome V2 capture and paired‐end sequenced on the Illumina platform (outsourced). The aim is to obtain 8.1 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA‐MEM algorithm (reference: http://bio‐bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A4GALT [Blood group, P1Pk system, P(2) phenotype], 111400 607922 101 100 100 99 [Blood group, P1Pk system, p phenotype], 111400 NOR polyagglutination syndrome, 111400 AAAS Achalasia‐addisonianism‐alacrimia syndrome, 231550 605378 73 100 100 100 AAGAB Keratoderma, palmoplantar, -

(12) Patent Application Publication (10) Pub. No.: US 2016/0281166 A1 BHATTACHARJEE Et Al
US 20160281 166A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0281166 A1 BHATTACHARJEE et al. (43) Pub. Date: Sep. 29, 2016 (54) METHODS AND SYSTEMIS FOR SCREENING Publication Classification DISEASES IN SUBJECTS (51) Int. Cl. (71) Applicant: PARABASE GENOMICS, INC., CI2O I/68 (2006.01) Boston, MA (US) C40B 30/02 (2006.01) (72) Inventors: Arindam BHATTACHARJEE, G06F 9/22 (2006.01) Andover, MA (US); Tanya (52) U.S. Cl. SOKOLSKY, Cambridge, MA (US); CPC ............. CI2O 1/6883 (2013.01); G06F 19/22 Edwin NAYLOR, Mt. Pleasant, SC (2013.01); C40B 30/02 (2013.01); C12O (US); Richard B. PARAD, Newton, 2600/156 (2013.01); C12O 2600/158 MA (US); Evan MAUCELI, (2013.01) Roslindale, MA (US) (21) Appl. No.: 15/078,579 (57) ABSTRACT (22) Filed: Mar. 23, 2016 Related U.S. Application Data The present disclosure provides systems, devices, and meth (60) Provisional application No. 62/136,836, filed on Mar. ods for a fast-turnaround, minimally invasive, and/or cost 23, 2015, provisional application No. 62/137,745, effective assay for Screening diseases, such as genetic dis filed on Mar. 24, 2015. orders and/or pathogens, in Subjects. Patent Application Publication Sep. 29, 2016 Sheet 1 of 23 US 2016/0281166 A1 SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS S{}}\\93? sau36 Patent Application Publication Sep. 29, 2016 Sheet 2 of 23 US 2016/0281166 A1 &**** ? ???zzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzz??º & %&&zzzzzzzzzzzzzzzzzzzzzzz &Sssssssssssssssssssssssssssssssssssssssssssssssssssssssss & s s sS ------------------------------ Patent Application Publication Sep. 29, 2016 Sheet 3 of 23 US 2016/0281166 A1 23 25 20 FG, 2. Patent Application Publication Sep. 29, 2016 Sheet 4 of 23 US 2016/0281166 A1 : S Patent Application Publication Sep. -

Blueprint Genetics Comprehensive Skeletal Dysplasias and Disorders
Comprehensive Skeletal Dysplasias and Disorders Panel Test code: MA3301 Is a 251 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of disorders involving the skeletal system. About Comprehensive Skeletal Dysplasias and Disorders This panel covers a broad spectrum of skeletal disorders including common and rare skeletal dysplasias (eg. achondroplasia, COL2A1 related dysplasias, diastrophic dysplasia, various types of spondylo-metaphyseal dysplasias), various ciliopathies with skeletal involvement (eg. short rib-polydactylies, asphyxiating thoracic dysplasia dysplasias and Ellis-van Creveld syndrome), various subtypes of osteogenesis imperfecta, campomelic dysplasia, slender bone dysplasias, dysplasias with multiple joint dislocations, chondrodysplasia punctata group of disorders, neonatal osteosclerotic dysplasias, osteopetrosis and related disorders, abnormal mineralization group of disorders (eg hypopohosphatasia), osteolysis group of disorders, disorders with disorganized development of skeletal components, overgrowth syndromes with skeletal involvement, craniosynostosis syndromes, dysostoses with predominant craniofacial involvement, dysostoses with predominant vertebral involvement, patellar dysostoses, brachydactylies, some disorders with limb hypoplasia-reduction defects, ectrodactyly with and without other manifestations, polydactyly-syndactyly-triphalangism group of disorders, and disorders with defects in joint formation and synostoses. Availability 4 weeks Gene Set Description -

Review of Genetics and Epidemiology. Charles Shaw
JMG Online First, published on November 21, 2005 as 10.1136/jmg.2005.038158 J Med Genet: first published as 10.1136/jmg.2005.038158 on 18 November 2005. Downloaded from Oesophageal atresia, tracheo-oesophageal fistula and the VACTERL association: review of genetics and epidemiology. Charles Shaw-Smith Department of Medical Genetics, Addenbrooke’s Hospital, Cambridge CB2 2QQ, Address for correspondence: Charles Shaw-Smith Department of Medical Genetics, Box 134, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 2QQ UK Telephone +44(0)1223 216446 Fax +44(0)1223 217054 Email [email protected] http://jmg.bmj.com/ Running title: Genetics of oesophageal atresia on September 26, 2021 by guest. Protected copyright. Copyright Article author (or their employer) 2005. Produced by BMJ Publishing Group Ltd under licence. J Med Genet: first published as 10.1136/jmg.2005.038158 on 18 November 2005. Downloaded from Abstract Oesophageal atresia and/or tracheo-oesophageal fistula are common malformations occurring in approximately 1 in 3500 births. In around half of cases (syndromic oesophageal atresia), there are other associated anomalies, with cardiac malformations being the most common. These may occur as part of the VACTERL association (OMIM 192350). In the remainder of cases, oesophageal atresia/tracheo-oesophageal fistula occur in isolation (non-syndromic oesophageal atresia). Data from twin and family studies suggest that, in the majority of cases, genetic factors do not play a major role, and yet there are well-defined instances of this malformation where genetic factors clearly are important. This is highlighted by the recent identification of no fewer than three separate genes with a role in the aetiology of oesophageal atresia. -

VACTERL/VATER Association Benjamin D Solomon
Solomon Orphanet Journal of Rare Diseases 2011, 6:56 http://www.ojrd.com/content/6/1/56 REVIEW Open Access VACTERL/VATER Association Benjamin D Solomon Abstract VACTERL/VATER association is typically defined by the presence of at least three of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities. In addition to these core component features, patients may also have other congenital anomalies. Although diagnostic criteria vary, the incidence is estimated at approximately 1 in 10,000 to 1 in 40,000 live-born infants. The condition is ascertained clinically by the presence of the above-mentioned malformations; importantly, there should be no clinical or laboratory-based evidence for the presence of one of the many similar conditions, as the differential diagnosis is relatively large. This differential diagnosis includes (but is not limited to) Baller-Gerold syndrome, CHARGE syndrome, Currarino syndrome, deletion 22q11.2 syndrome, Fanconi anemia, Feingold syndrome, Fryns syndrome, MURCS association, oculo-auriculo-vertebral syndrome, Opitz G/BBB syndrome, Pallister- Hall syndrome, Townes-Brocks syndrome, and VACTERL with hydrocephalus. Though there are hints regarding causation, the aetiology has been identified only in a small fraction of patients to date, likely due to factors such as a high degree of clinical and causal heterogeneity, the largely sporadic nature of the disorder, and the presence of many similar conditions. New genetic research methods offer promise that the causes of VACTERL association will be better defined in the relatively near future. Antenatal diagnosis can be challenging, as certain component features can be difficult to ascertain prior to birth. -

Chromosomal Microarray, Postnatal, Clarisure® Oligo-SNP
Diagnostic Services Pediatrics Test Summary Chromosomal Microarray, Postnatal, ClariSure® Oligo-SNP The American College of Medical Genetics (ACMG) Test Code: 16478(X) recommends CMA testing as a first-line genetic test for developmental delay, intellectual disability, ASDs, and Specimen Requirements: 10 mL room-temperature whole multiple congenital anomalies.2,4 This recommendation blood (sodium-heparin, green-top tube); 5 mL minimum is based, in part, on a literature review that included over 21,000 patients with developmental delay/intellectual CPT Code*: 81229 disability, ASDs, or multiple congenital anomalies. The diagnostic yield was 15% to 20% for CMA testing versus ~3% for G-banded karyotyping and ~6% for subtelomeric Clinical Use fluorescence in situ hybridization (FISH) in combination • Determine the genetic etiology of developmental with G-banded karyotyping.5 In individuals with complex delay, intellectual disability, autism spectrum disorders ASDs, CMA testing can result in a diagnostic yield of over (ASDs; pervasive developmental disorders), and 25%.2 ACMG still considers karyotyping a first-line test multiple congenital anomalies when patients are suspected of having a recognizable • Confirm or exclude the diagnosis of known chromosomal syndrome such as trisomy 21 or 18, Turner chromosomal syndromes syndrome, or Klinefelter syndrome.2,4 • Further define ambiguities arising from cytogenetic The oligonucleotide-single nucleotide polymorphism or FISH studies (oligo-SNP) array contains over 2.6 million probes and • Assist in clinical management and genetic counseling covers regions of known and likely CNVs. It can confirm the diagnosis of suspected disorders associated with known Clinical Background chromosomal syndromes and is especially well suited Global developmental delay, intellectual disability (mental for determining the genetic cause of less well-described retardation), ASDs, and multiple congenital anomalies may disorders. -

Wo 2009/039966 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 2 April 2009 (02.04.2009) WO 2009/039966 A2 (51) International Patent Classification: (74) Agent: ARTH, Hans-Lothar; ABK Patent Attorneys, Jas- A61K 38/17 (2006.01) A61P 11/00 (2006.01) minweg 9, 14052 Berlin (DE). A61K 38/08 (2006.01) A61P 25/28 (2006.01) A61P 31/20 (2006.01) A61P 31/00 (2006.01) (81) Designated States (unless otherwise indicated, for every A61P 3/00 (2006.01) A61P 35/00 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/00 (2006.01) A61P 37/00 (2006.01) AO, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY,BZ, CA, CH, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, (21) International Application Number: EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, PCT/EP2008/007500 IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY,MA, MD, ME, MG, MK, MN, MW, (22) International Filing Date: MX, MY,MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, 9 September 2008 (09.09.2008) RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY,TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (25) Filing Language: English ZW (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, 07017754.8 11 September 2007 (11.09.2007) EP ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AT,BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, (71) Applicant (for all designated States except US): MONDO- FR, GB, GR, HR, HU, IE, IS, IT, LT,LU, LV,MC, MT, NL, BIOTECH LABORATORIES AG [LLLI]; Herrengasse NO, PL, PT, RO, SE, SI, SK, TR), OAPI (BF, BJ, CF, CG, 21, FL-9490 Vaduz (LI). -

Early ACCESS Diagnosed Conditions List
Iowa Early ACCESS Diagnosed Conditions Eligibility List List adapted with permission from Early Intervention Colorado To search for a specific word type "Ctrl F" to use the "Find" function. Is this diagnosis automatically eligible for Early Medical Diagnosis Name Other Names for the Diagnosis and Additional Diagnosis Information ACCESS? 6q terminal deletion syndrome Yes Achondrogenesis I Parenti-Fraccaro Yes Achondrogenesis II Langer-Saldino Yes Schinzel Acrocallosal syndrome; ACLS; ACS; Hallux duplication, postaxial polydactyly, and absence of the corpus Acrocallosal syndrome, Schinzel Type callosum Yes Acrodysplasia; Arkless-Graham syndrome; Maroteaux-Malamut syndrome; Nasal hypoplasia-peripheral dysostosis-intellectual disability syndrome; Peripheral dysostosis-nasal hypoplasia-intellectual disability (PNM) Acrodysostosis syndrome Yes ALD; AMN; X-ALD; Addison disease and cerebral sclerosis; Adrenomyeloneuropathy; Siemerling-creutzfeldt disease; Bronze schilder disease; Schilder disease; Melanodermic Leukodystrophy; sudanophilic leukodystrophy; Adrenoleukodystrophy Pelizaeus-Merzbacher disease Yes Agenesis of Corpus Callosum Absence of the corpus callosum; Hypogenesis of the corpus callosum; Dysplastic corpus callosum Yes Agenesis of Corpus Callosum and Chorioretinal Abnormality; Agenesis of Corpus Callosum With Chorioretinitis Abnormality; Agenesis of Corpus Callosum With Infantile Spasms And Ocular Anomalies; Chorioretinal Anomalies Aicardi syndrome with Agenesis Yes Alexander Disease Yes Allan Herndon syndrome Allan-Herndon-Dudley -

Blueprint Genetics Congenital Structural Heart Disease Panel
Congenital Structural Heart Disease Panel Test code: CA1501 Is a 114 gene panel that includes assessment of non-coding variants. Is ideal for patients with congenital heart disease, particularly those with features of hereditary disorders. Is not ideal for patients suspected to have a ciliopathy or a rasopathy. For those patients, please consider our Primary Ciliary Dyskinesia Panel and our Noonan Syndrome Panel, respectively. About Congenital Structural Heart Disease There are many types of congenital heart disease (CHD) ranging from simple asymptomatic defects to complex defects with severe, life-threatening symptoms. CHDs are the most common type of birth defect and affect at least 8 out of every 1,000 newborns. Annually, more than 35,000 babies in the United States are born with CHDs. Many of these CHDs are simple conditions and need no treatment or are easily repaired. Some babies are born with complex CHD requiring special medical care. The diagnosis and treatment of complex CHDs has greatly improved over the past few decades. As a result, almost all children who have complex heart defects survive to adulthood and can live active, productive lives. However, many patients who have complex CHDs continue to need special heart care throughout their lives. In the United States, more than 1 million adults are living with congenital heart disease. Availability 4 weeks Gene Set Description Genes in the Congenital Structural Heart Disease Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ABL1 Congenital -

Imaging Casebook Feingold Syndrome: Microcephaly, Esophageal Atresia, Type III Laryngeal Cleft, Malrotation, Limb Anomalies
Imaging Casebook Feingold Syndrome: Microcephaly, Esophageal Atresia, Type III Laryngeal Cleft, Malrotation, Limb Anomalies Thomas E. Herman, MD nasogastric tube in the upper esophagus consistent with esophageal Marilyn J. Siegel, MD atresia without a distal fistula. Bronchoscopy and esophagoscopy demonstrated esophageal atresia without a proximal fistula and Journal of Perinatology (2004) 24, 568–570. doi:10.1038/sj.jp.7211144 also a type III laryngotracheoesophageal cleft. Subsequently, a gastrostomy tube was placed. An upper GI series (Figure 2) demonstrated malrotation. Therefore, a Ladd’s procedure and a CASE PRESENTATION cervical esophagostomy to manage secretions were performed. A 2085 g infant was born at 37 weeks gestation to a 36-year-old Microcephaly was present clinically and an MRI (Figure 3) gravida 2, para 1 mother. The pregnancy had been complicated by demonstrated a thin corpus callosum. Radiographs of the feet in polyhydramnios. The Apgar scores were 8 and 9 at 1 and 5 minutes the neonatal period and hand films obtained at 2 years of age of age. Postnatally, the infant became dusky with attempted (Figure 4) demonstrate shortening of all middle phalanges of the nasogastric tube placement. The initial radiography of the chest toes and slight shortening of the second and fifth middle phalanges and abdomen (Figure 1) demonstrated a gasless abdomen and a of the fingers. A cardiac sonogram demonstrated a large paramembranous VSD. The patient underwent dilatation of the esophageal pouch and primary reanastomosis of the esophagus. The type III laryngotracheoesophageal cleft was also repaired prior to the esophageal reanastomosis. Figure 1. Anteroposterior radiograph of the chest and upper abdomen at birth. -
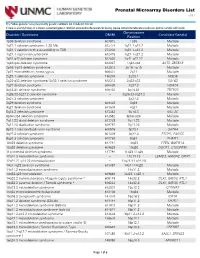
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome