SN1 Vs. E1 and the Gibbs-Helmholtz Equation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bsc Chemistry
Subject Chemistry Paper No and Title 05, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) Module No and Title 15, The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds Module Tag CHE_P5_M15 CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. NGP Participation 3.1 NGP by Heteroatom Lone Pairs 3.2 NGP by alkene 3.3 NGP by Cyclopropane, Cyclobutane or a Homoallyl group 3.4 NGP by an Aromatic Ring 4. Neighbouring Group Participation on SN2 Reactions 5. Neighbouring Group Participation on SN1 Reactions 6. Neighbouring Group and Rearrangement 7. Examples 8. Summary CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds 1. Learning Outcomes After studying this module, you shall be able to Know about NGP reaction Learn reaction mechanism of NGP reaction Identify stereochemistry of NGP reaction Evaluate the factors affecting the NGP reaction Analyse Phenonium ion, NGP by alkene, and NGP by heteroatom. 2. Introduction The reaction centre (carbenium centre) has direct interaction with a lone pair of electrons of an atom or with the electrons of s- or p-bond present within the parent molecule but these are not in conjugation with the reaction centre. A distinction is sometimes made between n, s and p- participation. An increase in rate due to Neighbouring Group Participation (NGP) is known as "anchimeric assistance". "Synartetic acceleration" happens to be the special case of anchimeric assistance and applies to participation by electrons binding a substituent to a carbon atom in a β- position relative to the leaving group attached to the α-carbon atom. -

Reaction Kinetics in Organic Reactions
Autumn 2004 Reaction Kinetics in Organic Reactions Why are kinetic analyses important? • Consider two classic examples in asymmetric catalysis: geraniol epoxidation 5-10% Ti(O-i-C3H7)4 O DET OH * * OH + TBHP CH2Cl2 3A mol sieve OH COOH5C2 L-(+)-DET = OH COOH5C2 * OH geraniol hydrogenation OH 0.1% Ru(II)-BINAP + H2 CH3OH P(C6H5)2 (S)-BINAP = P(C6 H5)2 • In both cases, high enantioselectivities may be achieved. However, there are fundamental differences between these two reactions which kinetics can inform us about. 1 Autumn 2004 Kinetics of Asymmetric Catalytic Reactions geraniol epoxidation: • enantioselectivity is controlled primarily by the preferred mode of initial binding of the prochiral substrate and, therefore, the relative stability of intermediate species. The transition state resembles the intermediate species. Finn and Sharpless in Asymmetric Synthesis, Morrison, J.D., ed., Academic Press: New York, 1986, v. 5, p. 247. geraniol hydrogenation: • enantioselectivity may be dictated by the relative reactivity rather than the stability of the intermediate species. The transition state may not resemble the intermediate species. for example, hydrogenation of enamides using Rh+(dipamp) studied by Landis and Halpern (JACS, 1987, 109,1746) 2 Autumn 2004 Kinetics of Asymmetric Catalytic Reactions “Asymmetric catalysis is four-dimensional chemistry. Simple stereochemical scrutiny of the substrate or reagent is not enough. The high efficiency that these reactions provide can only be achieved through a combination of both an ideal three-dimensional structure (x,y,z) and suitable kinetics (t).” R. Noyori, Asymmetric Catalysis in Organic Synthesis,Wiley-Interscience: New York, 1994, p.3. “Studying the photograph of a racehorse cannot tell you how fast it can run.” J. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

SN2 , SN1 , E2 , & E1: Substitution and Elimination Reactions
SN2 , SN1 , E2 , & E1: Substitution and Elimination Reactions • Nucleophilic Substitution Reactions (SN2 and SN1) replace a leaving group with a nucleophile (Nu: or Nu: - ) • Elimination Reactions (E2 and E1) generate a double bond by loss of " A+ " and " B: - " • They may compete with each other Nucleophilic Substitution Reactions - SN2 Reaction: • Reaction is: o Stereospecific (Walden Inversion of configuration) o Concerted - all bonds form and break at same time o Bimolecular - rate depends on concentration of both nucleophile and substrate • Substrate: o Best if primary (one substituent on carbon bearing leaving group) o works if secondary, fails if tertiary • Nucleophile: o Best if more reactive (i.e. more anionic or more basic) • Leaving Group: Best if more stable (i.e. can support negative charge well): o TsO- (very good) > I- > Br- > Cl- > F- (poor) o RF , ROH , ROR , RNH2 are NEVER Substrates for SN2 reactions o Leaving Groups on double-bonded carbons are never replaced by SN2 reactions • Solvent: Polar Aprotic (i.e. no OH) is best. o For example dimethylsulfoxide ( CH3SOCH3 ), dimethylformamide ( HCON(CH3)2 ), acetonitrile ( CH3CN ). o Protic solvents (e.g. H2O or ROH) deactivate nucleophile by hydrogen bonding but can be used in some case Intro Chem Handouts Substitution & Elimination Reactions Page 1 of 3 Nucleophilic Substitution Reactions – SN1 Reaction: • Reaction is: o Non-stereospecific (attack by nucleophile occurs from both sides) o Non-concerted - has carbocation intermediate o Unimolecular - rate depends on concentration of only the substrate • Substrate: o Best if tertiary or conjugated (benzylic or allylic) carbocation can be formed as leaving group departs o never primary • Nucleophile: o Best if more reactive (i.e. -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

SUBSTITUTION and ELIMINATION REACTIONS Nucleophilic Substitution at Sp3 Carbon Substitution Is the Replacement of One Group by Another
SUBSTITUTION AND ELIMINATION REACTIONS Nucleophilic Substitution at sp3 Carbon Substitution is the replacement of one group by another. The conversion of an alkyl halide into an alcohol is one of the most widely studied substitution reactions. _ _ OH R-X R-OH + X + OH- is called nucleophile and X- is called leaving group. A nucleophile is a reagent that brings an electron pair. Kinetic measurements on the reactions of alkyl halides with a number of different nucleophile show two extreme types. In the one type, Rate = k1[R-X] (1) And in the second type Rate = k2[R-X][Nu:] (2) In some cases the rate equations are found to be mixed or are complicated due to other reasons. Mechanisms The most common mechanisms of substitution reactions are SN1 and SN2 mechanisms. SN1 mechanism follows rate equation (1), i.e. first order kinetics while SN2 mechanism follows rate equation (2), i.e. second order kinetics. The SN2 Mechanism SN2 stands for substitution nucleophilic bimolecular. In this mechanism the nucleophile approaches the substrate from a position 180º away from the leaving group (backside attack). The reaction is a one step process with no intermediate. _ _ Y + C X Y C X Y C + X The C-Y bond is formed as the C-X bond is broken. The energy required to break the C-X bond is supplied by the simultaneous formation of the C-Y bond. During the formation of the transition state the hybridization of the carbon atom changes from sp3 to sp2 with an approximately perpendicular p-orbital. -

Carbocation Catalysis for Organic Synthesis Shengjun Ni LIC
Carbocation Catalysis for Organic Synthesis Shengjun Ni Doctoral Thesis LIC OR DOC Thesis Stockholm 201 year8 Akademisk avhandling som med tillstånd av Kungliga Tekniska Högskolan i Stockholm framlägges till offentlig granskning för avläggande av doktorsexamen i kemi med inriktning mot organisk kemi fredagen den 26 oktober kl 14.00 i sal F3, KTH, Lindstedtsvägen 26, Stockholm. Avhandlingen försvaras på engelska. Opponent är professor Bernd Plietker, Universität Stuttgart, Germany. ISBN 978-91-7729-966-0 ISSN 1654-1081 TRITA-CBH-FOU-2018:44 © Shengjun Ni, 2018 Universitetsservice US AB, Stockholm II Shengjun Ni, 2018: “Carbocation Catalysis for Organic Synthesis”, KTH Engineering Sciences in Chemistry, Biotechnology and Health, Royal Institute of Technology, SE-100 44 Stockholm, Sweden. Abstract The most common view of carbocations in organic chemistry is that they are short-lived intermediates in several fundamental reactions, e.g. the classic SN1-reaction. However, carbocations that can delocalize their positive charge can be stable enough to be isolated and used as Lewis acid catalysts, phase transfer catalysts or oxidants in various reactions. The theme of this thesis concerns applying trityl cations as Lewis acid catalysts in different organic reactions. The first chapter presents a general introduction of the field of Lewis acids, the characteristics and applications of carbocations in different organic reactions, and the aims of this thesis. The second chapter describes the carbocation-catalyzed asymmetric Diels– Alder reactions assisted by chiral counteranions. The third chapter shows that carbocations can be utilized as catalysts in oxa-Diels–Alder reactions with unactivated aldehydes and dienes as substrates. The fourth chapter investigates the application of carbocation catalysis in bromination reactions for selective functionalization at the benzylic position and on the aromatic ring, respectively. -

Transition-Metal Catalysis of Nucleophilic Substitution Reactions: a Radical Alternative to SN1 and SN2 Processes Gregory C
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Outlook http://pubs.acs.org/journal/acscii Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes Gregory C. Fu* Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States ABSTRACT: Classical methods for achieving nucleophilic substitutions of alkyl electrophiles (SN1 and SN2) have limited scope and are not generally amenable to enantioselective variants that employ readily available racemic electrophiles. Radical-based pathways catalyzed by chiral transition-metal complexes provide an attractive approach to addressing these limitations. ■ UNSOLVED CHALLENGES IN NUCLEOPHILIC SUBSTITUTION REACTIONS OF ALKYL ELECTROPHILES Nucleophilic substitution of an alkyl electrophile is an extremely useful strategy in organic synthesis (Figure 1). SN1 and SN2 reactions are the two classical pathways for achieving this process; both have significant limitations.1,2 Figure 2. Limitations of the SN1 reaction. nucleophile and the electrophile, and it is therefore often unsuccessful in the case of hindered primary (e.g., neopentyl) Figure 1. Nucleophilic substitution reactions of alkyl electrophiles: and secondary electrophiles (Figure 3). Furthermore, SN2 SN1 and SN2 reactions. reactions are generally conducted under Brønsted-basic conditions, which can -

Aliphatic Nucleophilic Substitution
Richard F. Daley and Sally J. Daley www.ochem4free.com Organic Chemistry Chapter 12 Aliphatic Nucleophilic Substitution 12.1 Naming Single Bonded Heteroatom Functional Groups 579 12.2 Comparing Nucleophilic Substitution Reaction Mechanisms 586 12.3 The SN1 and SN2 Reaction Mechanisms 588 12.4 Stereochemistry of Nucleophilic Substitutions 592 12.5 The Substrate 595 12.6 Nucleophiles and Leaving Groups 601 12.7 Common Nucleophiles 606 12.8 The Reaction Medium 607 12.9 SN1 versus SN2 613 12.10 Halide Nucleophiles 613 Synthesis of 1-Bromobutane 616 12.11 Oxygen Nucleophiles 620 12.12 Nitrogen Nucleophiles 625 Synthesis of 2,5-Diaminoadipic Acid 628 12.13 Carbon Nucleophiles 631 12.14 Neighboring Group Participation 634 Special Topic - SN1 vs. SN2 637 Key Ideas from Chapter 12 641 Organic Chemistry - Ch 12 578 Daley & Daley Copyright 1996-2005 by Richard F. Daley & Sally J. Daley All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior written permission of the copyright holder. www.ochem4free.com 5 July 2005 Organic Chemistry - Ch 12 579 Daley & Daley Chapter 12 Aliphatic Nucleophilic Substitution Chapter outline 12.1 Naming Single Bonded Heteroatom Functional Groups Naming alcohols, ethers, phenols, thiols, thioethers, sulfides, and amines 12.2 Comparing Nucleophilic Substitution Reaction Mechanisms A comparison of the mechanism for SN1 and SN2 reactions with the mechanism of -

1 Msc(Chem) 1St Sem Dr Sanjeev Kumar Jha M.L.T. College, Saharsa Reaction Mechanism: Structure and Reactivity Types Of
MSc(Chem) 1st Sem Dr Sanjeev Kumar Jha M.L.T. College, Saharsa Reaction Mechanism: Structure and reactivity Types of reactions:-- 1.Addition reaction An addition reaction , is simply termed as a reaction where two or more molecules combine to form a larger one (the adduct). Addition reactions are limited to chemical compounds that have multiple bonds, such as molecules with carbon–carbon double bonds (alkenes), or with triple bonds (alkynes), and compounds that have rings, which are also considered points of unsaturation. Molecules containing carbon— hetero double bonds like carbonyl (C=O) groups, or imine (C=N) groups, can undergo addition, as they too have double-bond character. An addition reaction is the reverse of an elimination reaction. For instance, the hydration of an alkene to an alcohol is reversed by dehydration. There are two main types of polar addition reactions: electrophilic addition and nucleophilic addition. Two non-polar addition reactions exist as well, called free-radical addition and cycloadditions. Addition reactions are also encountered in polymerizations and called addition polymerization. 1 General overview of addition reactions. Top to bottom: electrophilic addition to alkene, nucleophilic addition of nucleophile to carbonyl and free-radical addition of halide to alkene. 2.Elimination reaction An elimination reaction is a type of reaction in which two substituents are removed from a molecule in either a one or two-step mechanism. The one- step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). -
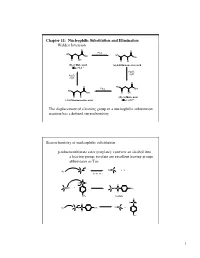
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

Chem 232 Lecture 28.Pdf
CHEM 232 University of Illinois Organic Chemistry I at Chicago UIC Lecture 28 Organic Chemistry 1 Professor Duncan Wardrop April 22, 2010 1 Today’s Lecture Topics Covered: 1. Aryl Halides - Bonding, Physical Properties and Reactions 2. Nucleophilic Aromatic Substitution of Chlorobenzene 3. Nucleophilic Aromatic Substitution: Addition-Elimination 4. Floxcin - Application of Nucleophilic Aromatic Substitution 5. Nucleophilic Aromatic Substitution: Elimination-Addition 2 What’s the Difference Between Ar- and Ph-? Phenyl refers specifically to this: Aryl is a general term for all aromatic ring systems: C N N O 3 Chapter 23 Aryl Halides 4 23.1 Bonding in Aryl Halides 5 Aryl Halides X Aryl halides are halides in which the halogen is attached directly to an aromatic ring. Carbon-halogen bonds in aryl halides are shorter and stronger than carbon- halogen bonds in alkyl halides. 6 Dissociation Energies of Selected Compounds Bond Energy: kJ/mol (kcal/mol) X = H X = Cl CH3CH2X sp3 410 (98) 339 (81) 2 H2C CHX sp 452 (108) 368 (88) X sp2 469 (112) 406 (97) 7 Resonance Picture X X X H H C-X bonds in aryl halides have more double bond character than C-X bonds in alkyl halides 8 23.2 Sources of Aryl Halides 9 Preparation of Aryl Halides Halogenation of arenes (Section 12.5) H Br FeBr3 Br2 HBr electrophilic aromatic substitution 10 Preparation of Aryl Halides The Sandmeyer reaction (Section 22.17) N H H N N Cl Cl NaNO2 CuCl O HCl, H2O O heat O N+ N+ N+ O– O– O– Primary Aryl Diazonium Aryl Chloride Arylamine Salt diazotization-nucleophilic aromatic substitution 11 Preparation of Aryl Halides The Schiemann reaction (Section 22.17) N H H N BF4 N F 1.