A Meta-Analysis of Alzheimer's Disease Brain Transcriptomic Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony
Supplementary Data Th2 and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in UBIOPRED Chih-Hsi Scott Kuo1.2, Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony Rowe3, Iaonnis Pandis2, Ana Sousa4, Julie Corfield5, Ratko Djukanovic6, Rene 7 7 8 2 1† Lutter , Peter J. Sterk , Charles Auffray , Yike Guo , Ian M. Adcock & Kian Fan 1†* # Chung on behalf of the U-BIOPRED consortium project team 1Airways Disease, National Heart & Lung Institute, Imperial College London, & Biomedical Research Unit, Biomedical Research Unit, Royal Brompton & Harefield NHS Trust, London, United Kingdom; 2Department of Computing & Data Science Institute, Imperial College London, United Kingdom; 3Janssen Research and Development, High Wycombe, Buckinghamshire, United Kingdom; 4Respiratory Therapeutic Unit, GSK, Stockley Park, United Kingdom; 5AstraZeneca R&D Molndal, Sweden and Areteva R&D, Nottingham, United Kingdom; 6Faculty of Medicine, Southampton University, Southampton, United Kingdom; 7Faculty of Medicine, University of Amsterdam, Amsterdam, Netherlands; 8European Institute for Systems Biology and Medicine, CNRS-ENS-UCBL, Université de Lyon, France. †Contributed equally #Consortium project team members are listed under Supplementary 1 Materials *To whom correspondence should be addressed: [email protected] 2 List of the U-BIOPRED Consortium project team members Uruj Hoda & Christos Rossios, Airways Disease, National Heart & Lung Institute, Imperial College London, UK & Biomedical Research Unit, Biomedical Research Unit, Royal -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

New Tests 2 Test Updates 4 CPT Code Updates 11 Deleted Tests 11 Diagnostics Update Volume XXI, No
Volume XXI, No. 6 JUNE 2021 MONTHLY TEST UPDATES Diagnostics Update New Tests 2 Test Updates 4 CPT Code Updates 11 Deleted Tests 11 Diagnostics Update Volume XXI, No. 6 | JUNE 2021 New Tests Use Anti-DFS70 antibodies may help identify individuals who do not have an Anti-Carbamylated Protein (CarP) Antibody 520311 ANA-associated Autoimmune Rheumatic Disease (AARD) especially in the absence of significant clinical findings.1 Anti-DFS70 Ab, especially when positive CPT 83516 in isolation (i.e. in the absence of AARD-associated autoantibodies), may Synonyms Anti-CarP antigen antibody; RA marker prevent unnecessary referrals and examinations of ANA-positive individuals.2 Special Instructions This test has not been approved for NY state clients. Limitations This test should be used with clinical findings and other Specimen Serum autoimmune testing; it cannot be used alone to rule out autoimmune disease. Volume 1 mL This test was developed and its performance characteristics determined Minimum Volume 0.5 mL by Labcorp. It has not been cleared or approved by the Food and Drug Container Red-top tube; serum from red-top tube; serum from a gel tube; or Administration. serum gel tube Methodology Enzyme-linked immunosorbent assay (ELISA) Collection Separate serum from cells within one hour of collection. Transfer to a Additional Information Anti-DFS70 antibodies target the dense fine speckled plastic transport tube before shipping. protein of 70 kDa which is identical to Lens Epithelium-Derived Growth Factor Storage Instructions Refrigerate or freeze. or transcription co-activator p75 (LEDGFp75). They are detectable in 2% to 22% Stability of healthy individuals and in less than 1% of patients with AARD are of unknown Temperature Period clinical significance. -

ELMO1 Signaling Is a Promoter of Osteoclast Function and Bone Loss ✉ Sanja Arandjelovic 1 , Justin S
ARTICLE https://doi.org/10.1038/s41467-021-25239-6 OPEN ELMO1 signaling is a promoter of osteoclast function and bone loss ✉ Sanja Arandjelovic 1 , Justin S. A. Perry 1, Ming Zhou 2, Adam Ceroi3, Igor Smirnov4, Scott F. Walk1, Laura S. Shankman1, Isabelle Cambré3, Suna Onengut-Gumuscu 5, Dirk Elewaut 3, Thomas P. Conrads 2 & ✉ Kodi S. Ravichandran 1,3 Osteoporosis affects millions worldwide and is often caused by osteoclast induced bone loss. 1234567890():,; Here, we identify the cytoplasmic protein ELMO1 as an important ‘signaling node’ in osteoclasts. We note that ELMO1 SNPs associate with bone abnormalities in humans, and that ELMO1 deletion in mice reduces bone loss in four in vivo models: osteoprotegerin deficiency, ovariectomy, and two types of inflammatory arthritis. Our transcriptomic analyses coupled with CRISPR/Cas9 genetic deletion identify Elmo1 associated regulators of osteoclast function, including cathepsin G and myeloperoxidase. Further, we define the ‘ELMO1 inter- actome’ in osteoclasts via proteomics and reveal proteins required for bone degradation. ELMO1 also contributes to osteoclast sealing zone on bone-like surfaces and distribution of osteoclast-specific proteases. Finally, a 3D structure-based ELMO1 inhibitory peptide reduces bone resorption in wild type osteoclasts. Collectively, we identify ELMO1 as a signaling hub that regulates osteoclast function and bone loss, with relevance to osteoporosis and arthritis. 1 Center for Cell Clearance, Department of Microbiology, Immunology, and Cancer Biology and Carter Immunology Center, University of Virginia, Charlottesville, VA, USA. 2 Inova Schar Cancer Institute, Inova Center for Personalized Health, Fairfax, VA, USA. 3 Inflammation Research Centre, VIB, and the Department of Biomedical Molecular Biology, Ghent Univeristy, Ghent, Belgium. -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/). -

Kidney V-Atpase-Rich Cell Proteome Database
A comprehensive list of the proteins that are expressed in V-ATPase-rich cells harvested from the kidneys based on the isolation by enzymatic digestion and fluorescence-activated cell sorting (FACS) from transgenic B1-EGFP mice, which express EGFP under the control of the promoter of the V-ATPase-B1 subunit. In these mice, type A and B intercalated cells and connecting segment principal cells of the kidney express EGFP. The protein identification was performed by LC-MS/MS using an LTQ tandem mass spectrometer (Thermo Fisher Scientific). For questions or comments please contact Sylvie Breton ([email protected]) or Mark A. Knepper ([email protected]). -

Elucidating the Molecular Mechanisms Involved in Assembly/Folding and Targeting V-Atpase A-Subunit Isoforms to Their Functional Destinations
Elucidating the Molecular Mechanisms Involved in Assembly/Folding and Targeting V-ATPase a-subunit Isoforms to their Functional Destinations By Sally Esmail A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Faculty of Dentistry University of Toronto © Copyright by Sally Esmail 2017 ii Elucidating the Molecular Mechanisms Involved in Assembly/Folding and Targeting V-ATPase a-subunit Isoforms to their Functional Destinations Sally Esmail Doctor of Philosophy Faculty of Dentistry University of Toronto 2017 Abstract Vacuolar H+-ATPases (V-ATPases) are proton pumps distributed across membranes of specialized cells and luminal compartments. V-ATPases in the plasma membrane of osteoclasts are responsible for acidifying the surface of bone, essential for bone resorption. V-ATPases in the plasma membrane of metastatic cells acidify the extracellular space to facilitate invasion. The V-ATPase a subunit has four isoforms (a1-a4) that localize to distinct compartments. In invasive cancer, plasma membrane expression of a3 and a4 are required for metastasis while a3 is specific for the plasma membrane of osteoclasts. Thus, both isoforms are potential therapeutic targets. Sequences of a isoforms reveal putative N-glycosylation sites within extracellular loop II (ELII). Upon PNGase F and Endo H treatment, all a isoforms showed faster mobility on SDS- PAGE indicating the presence of N-linked oligosaccharide. Using site-directed mutagenesis, I showed that deglycosylated a1–a4 had shorter half-lifes, more rapid proteasomal degradation, endoplasmic reticulum (ER) retention, defective Golgi trafficking, and an inability to associate with ER assembly factor, VMA21. In addition, deglycosylated a4 showed defective cell-surface iii expression and assembly. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -
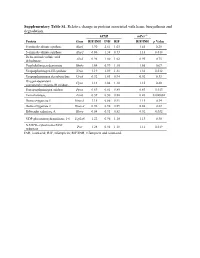
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-