Rev Iss Web Nph 12790 203-1 22..28
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 4. Experimental Pollinations Within and Between Members of the Drosera Peltata Complex (Droseraceae)
Chapter 4. Experimental pollinations within and between members of the Drosera peltata complex (Droseraceae) 4.1 Introduction The study of plant breeding systems has taxonomic and evolutionary implications. Inter-taxon compatibility, measured by seed set following cross pollinations, has been used to define taxon limits. Individuals that are cross compatible and produce fertile offspring are considered to be conspecific, or belong to the same species, whereas those that do not are considered separate species. This is known as the biological species concept (Mayr and Ashlock 1991~ Mayr 1993). The phylogenetic species concept approach also defines taxa by their gene flow, and estimates evolutionary relationships between entities. It is considered that unimpeded gene flow occurs within species, but that reproductive barriers prevent gene flow between species (Mayr 1993). The nature and extent of these reproductive barriers are considered to be commensurate in scale to the evolutionary divergence between the taxa of interest (Nixon and Wheeler 1990). Reproductive isolation mechanisms may exist that prevent gene flow between indi vidual plants or plant populations. These mechanisms may operate prior to pollination, between pollination and fertilization or post-fertilisation (Levin 1971). For example, otherwise reproductively compatible taxa may grow allopatrically, or may be sympatric but with non-overlapping flowering seasons, which prevents gene flow. Sympatlic congeners may also have floral morphologies that allows access by some pollinators but not others, preventing heterospecific pollen transfer. Prezygotic reproductive barriers prevent heterospecific pollen from germinating on the stigmas, or chemically inhibit pollen tube growth in the style. Postzygotic reproductive barriers may occur so that hybrid zygotes fail to develop. -

Status of Insectivorous Plants in Northeast India
Technical Refereed Contribution Status of insectivorous plants in northeast India Praveen Kumar Verma • Shifting Cultivation Division • Rain Forest Research Institute • Sotai Ali • Deovan • Post Box # 136 • Jorhat 785 001 (Assam) • India • [email protected] Jan Schlauer • Zwischenstr. 11 • 60594 Frankfurt/Main • Germany • [email protected] Krishna Kumar Rawat • CSIR-National Botanical Research Institute • Rana Pratap Marg • Lucknow -226 001 (U.P) • India Krishna Giri • Shifting Cultivation Division • Rain Forest Research Institute • Sotai Ali • Deovan • Post Box #136 • Jorhat 785 001 (Assam) • India Keywords: Biogeography, India, diversity, Red List data. Introduction There are approximately 700 identified species of carnivorous plants placed in 15 genera of nine families of dicotyledonous plants (Albert et al. 1992; Ellison & Gotellli 2001; Fleischmann 2012; Rice 2006) (Table 1). In India, a total of five genera of carnivorous plants are reported with 44 species; viz. Utricularia (38 species), Drosera (3), Nepenthes (1), Pinguicula (1), and Aldrovanda (1) (Santapau & Henry 1976; Anonymous 1988; Singh & Sanjappa 2011; Zaman et al. 2011; Kamble et al. 2012). Inter- estingly, northeastern India is the home of all five insectivorous genera, namely Nepenthes (com- monly known as tropical pitcher plant), Drosera (sundew), Utricularia (bladderwort), Aldrovanda (waterwheel plant), and Pinguicula (butterwort) with a total of 21 species. The area also hosts the “ancestral false carnivorous” plant Plumbago zelayanica, often known as murderous plant. Climate Lowland to mid-altitude areas are characterized by subtropical climate (Table 2) with maximum temperatures and maximum precipitation (monsoon) in summer, i.e., May to September (in some places the highest temperatures are reached already in April), and average temperatures usually not dropping below 0°C in winter. -
A Biological Survey of the Southern Mount Lofty Ranges
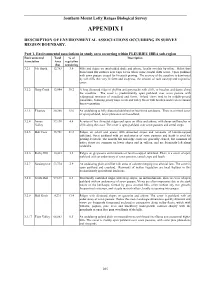
Southern Mount Lofty Ranges Biological Survey APPENDIX I DESCRIPTION OF ENVIRONMENTAL ASSOCIATIONS OCCURRING IN SURVEY REGION BOUNDARY. Part 1. Environmental associations in study area occurring within FLEURIEU IBRA sub-region Environmental Total % of Description Association Area vegetation (ha) remaining 3.2.1 Mt. Rapid 12,763 3.9 Hills and ridges on interbedded shale and arkose, locally overlain by tillite. Relict fans form broad flat surfaces near Cape Jervis where some coastal cliffs occur. Open parkland with sown pasture is used for livestock grazing. The scenery of the coastline is dominated by tall cliffs that vary in form and steepness, the amount of rock outcrop and vegetative cover. 3.2.2 Deep Creek 12,984 30.2 A long dissected ridge of phyllite and greywacke with cliffs, or beaches and dunes along the coastline. The cover is predominantly open parkland over sown pasture with widespread remnants of woodland and forest. Inland views tend to be middle-ground panoramic, featuring grassy ridge crests and valley floors with bracken and reed or remnant forest vegetation. 3.2.3 Fleurieu 30,389 15.6 An undulating to hilly dissected tableland on lateritized sandstone. There is a mixed cover of open parkland, forest plantation and woodland. 3.2.4 Inman 37,130 4.4 A series of low dissected ridges and spurs on tillite and arkose, with dunes and beaches or Valley cliffs along the coast. The cover is open parkland over sown pastures and cereal crops. 3.2.5 Bob Tiers 15,761 21.3 Ridges on schist and gneiss with dissected slopes and remnantsof laterite-capped tableland. -

Carnivorous Plant Responses to Resource Availability
Carnivorous plant responses to resource availability: environmental interactions, morphology and biochemistry Christopher R. Hatcher A doctoral thesis submitted in partial fulfilment of requirements for the award of Doctor of Philosophy of Loughborough University November 2019 © by Christopher R. Hatcher (2019) Abstract Understanding how organisms respond to resources available in the environment is a fundamental goal of ecology. Resource availability controls ecological processes at all levels of organisation, from molecular characteristics of individuals to community and biosphere. Climate change and other anthropogenically driven factors are altering environmental resource availability, and likely affects ecology at all levels of organisation. It is critical, therefore, to understand the ecological impact of environmental variation at a range of spatial and temporal scales. Consequently, I bring physiological, ecological, biochemical and evolutionary research together to determine how plants respond to resource availability. In this thesis I have measured the effects of resource availability on phenotypic plasticity, intraspecific trait variation and metabolic responses of carnivorous sundew plants. Carnivorous plants are interesting model systems for a range of evolutionary and ecological questions because of their specific adaptations to attaining nutrients. They can, therefore, provide interesting perspectives on existing questions, in this case trait-environment interactions, plant strategies and plant responses to predicted future environmental scenarios. In a manipulative experiment, I measured the phenotypic plasticity of naturally shaded Drosera rotundifolia in response to disturbance mediated changes in light availability over successive growing seasons. Following selective disturbance, D. rotundifolia became more carnivorous by increasing the number of trichomes and trichome density. These plants derived more N from prey and flowered earlier. -

Florida Council of Bromeliad Societies, Inc
Florida Council of Bromeliad Societies, Inc. In This Issue: 2007 Shows and Sales Cold Hardy Bromeliads List Vol. 27 Issue 1 February 2007 FCBS Affiliated Societies and Representatives B. Guild Tampa Bay Caloosahatchee Tom Wolfe Vicky Chirnside 5211 Lake LeClare Road 951 Southland Road Lutz 33558 Venice 34293 813-961-1475 941-493-5825 [email protected] [email protected] Bob Teems Tom Foley 813-855-0938 239-458-4656 Broward County Fl. East Coast Jose Donayre Calandra Thurrott 1240 Jefferson St. 713 Breckenridge Drive Hollywood 33019-1807 Port Orange 32127 954-925-5112 386-761-4804 Jcadonayre @bellsouth.net [email protected] Colleen Hendrix Carolyn Schoenau 954-530-0076 352-372-6589 Central Florida F. West Coast Betsy McCrory Linda Sheetz 3615 Boggy Creek Rd. 1153 Williams Dr. S Kissimmee 34744 St. Petersburg 33705 407-348-2139 727-864-3165 [email protected] [email protected] Butch Force Brian Corey 407-886-4814 727-864-3165 South Florida Gainesville Juan Espinosa-Almodovar Al Muzzell P.O. Box 430722 P.O. Box 14442 Miami 33243 Gainesville 32604 305-667-6155 352-372-4576 [email protected] John R. Moxley Michael Michalski 352-528-0783 305-279-2416 (Continued on the inside back cover.) 2007 Bromeliad Extravaganza Presented by Florida Council of Bromeliad Societies Hosted by the Bromeliad Society of Broward County Saturday, September 29, 2007 at the Hilton Ft. Lauderdale Airport Hotel 1870 Griffin Rd. Dania Beach, FL 33004 954-920-3300 954-920-3348 (fax) Room rates: Single or double $89.00 Rates in effect until September 14, 2007 Sale, Banquet, Raffle and Rare Plant Auction will take place at the same location. -

Geobio-Center LMU Report 2014 / 2015
GeoBio-Center LMU LMU ReportGeoBio-Center 2014 / 2015 Report 2012 / 2013 GeoBio-CenterLMU Report 2014 / 2015 Editor: Dirk Erpenbeck, Angelo Poliseno Layout: Lydia Geißler Cover composition: Lydia Geißler GeoBio-Center LMU, Richard-Wagner-Str. 10, 80333 München http://www.geobio-center.uni-muenchen.de Contents Welcoming note ......................................................................................................................4 Achievements of the GeoBio-Center LMU members 2014 & 2015 at a glance ......................5 Members of the GeoBio-CenterLMU ........................................................................................6 Memorial to Alexander Volker Altenbach (1953-2015) ..........................................................9 Publications in ISI-indexed Journals ...................................................................................14 Other peer-reviewed Publications .......................................................................................21 Further Publications .............................................................................................................23 Grants and Stipends ............................................................................................................26 Honors and Awards ..............................................................................................................27 Presentations on Conferences and Symposia ....................................................................28 Teaching ................................................................................................................................35 -

Towards Resolving Lamiales Relationships
Schäferhoff et al. BMC Evolutionary Biology 2010, 10:352 http://www.biomedcentral.com/1471-2148/10/352 RESEARCH ARTICLE Open Access Towards resolving Lamiales relationships: insights from rapidly evolving chloroplast sequences Bastian Schäferhoff1*, Andreas Fleischmann2, Eberhard Fischer3, Dirk C Albach4, Thomas Borsch5, Günther Heubl2, Kai F Müller1 Abstract Background: In the large angiosperm order Lamiales, a diverse array of highly specialized life strategies such as carnivory, parasitism, epiphytism, and desiccation tolerance occur, and some lineages possess drastically accelerated DNA substitutional rates or miniaturized genomes. However, understanding the evolution of these phenomena in the order, and clarifying borders of and relationships among lamialean families, has been hindered by largely unresolved trees in the past. Results: Our analysis of the rapidly evolving trnK/matK, trnL-F and rps16 chloroplast regions enabled us to infer more precise phylogenetic hypotheses for the Lamiales. Relationships among the nine first-branching families in the Lamiales tree are now resolved with very strong support. Subsequent to Plocospermataceae, a clade consisting of Carlemanniaceae plus Oleaceae branches, followed by Tetrachondraceae and a newly inferred clade composed of Gesneriaceae plus Calceolariaceae, which is also supported by morphological characters. Plantaginaceae (incl. Gratioleae) and Scrophulariaceae are well separated in the backbone grade; Lamiaceae and Verbenaceae appear in distant clades, while the recently described Linderniaceae are confirmed to be monophyletic and in an isolated position. Conclusions: Confidence about deep nodes of the Lamiales tree is an important step towards understanding the evolutionary diversification of a major clade of flowering plants. The degree of resolution obtained here now provides a first opportunity to discuss the evolution of morphological and biochemical traits in Lamiales. -

Carnivorous Plant Newsletter V44 N4 December 2015
Technical Refereed Contribution Several pygmy Sundew species possess catapult-flypaper traps with repetitive function, indicating a possible evolutionary change into aquatic snap traps similar to Aldrovanda Siegfried R. H. Hartmeyer and Irmgard Hartmeyer • Weil am Rhein • Germany • s.hartmeyer@ t-online.de • www.hartmeyer.de Keywords: Drosera, pygmy Sundew, Aldrovanda, Dionaea, Droseraceae, Collembola, carnivorous plant, catapult-flypaper trap, snap trap, snap-tentacle, functional morphology, phylogeny. Abstract: Approximately 50 species of pygmy Sundews (genus Drosera, section Bryastrum) occur in the South of Australia and one each in New Zealand (D. pygmaea) and Venezuela (D. meristo- caulis). They grow mainly as small stemless rosettes possessing minute trapping leaves of 1-2 mm diameter with prominent marginal tentacles, or have elongated erect stems. The caulescent species possess only mucus-producing tentacles that are most effective in capturing small flying insects. The acaulescent species in contrast are specialized on crawling prey (Verbeek & Boasson 1993) and have developed mucus-free snap-tentacles (Fig. 1), able to bend surprisingly rapidly towards the leaf center. They lift prey like, e.g. springtails (Collembola) from the ground and carry it with a 180°-movement from the periphery of the plant onto the sticky leaf. Our examinations brought to light that several small species of section Bryastrum are able to catapult small animals even within fractions of a second. If the whole leaf is touched, several or even all marginal tentacles perform such bending movements simultaneously. We documented this behavior on video, featured on our film “Catapults in Pygmyland” on YouTube (www.youtube.com/watch?v=5k7GYGibdjM). Our results prove that more than only one species in the genus Drosera possess rapidly moving catapult-flypaper traps and that the examined pygmy catapults show a further specialization and function repeatedly (in contrast to the one-shot snap tentacles of D. -

Carnivorous Plant Newsletter Vol 48, No 2, June 2019
Byblis in cultivation in the tropics and in temperate climates Gregory Allan • Birmingham • United Kingdom • [email protected] Cindy Chiang • Singapore • [email protected] This article has been written based mostly on the authors’ experiences in growing Byblis in the UK and in Singapore. It is hoped that growers in regions with different climates will be able to extrapo- late from the information provided here, adjusting the methods described below to suit their own growing conditions. Hopefully much of the information provided here is universal in its application. Introduction If any genus of carnivorous plants deserves to be better known, both by horticulturalists and botanists, it is Byblis. The common name (ironically rarely used by enthusiasts) for the genus is “rainbow plants”, on account of the prismatic effect that can be produced when the sun shines on their copiously-produced sticky glands. All species follow a basic morphological plan: they have a central stem from which radiate numerous filiform leaves and scapes with showy flowers that typi- cally have purple petals (although white forms of most species are known) and vivid yellow anthers. Virtually all parts of the plants are covered in mucilage-tipped stalked glands (colloquially referred to as “tentacles”), which efficiently capture small insects, as well as microscopic sessile digestive glands. Another interesting characteristic of the genus is the presence of pulvini in most, if not all, species. Pulvini are swellings at the base of pedicels which, usually after successful pollination, uti- lize hydraulics to bend the pedicel, and consequently the developing fruit, downwards towards the ground. This phenomenon was discovered by Brian Barnes in the early 21st century. -

Southern Gulf, Queensland
Biodiversity Summary for NRM Regions Species List What is the summary for and where does it come from? This list has been produced by the Department of Sustainability, Environment, Water, Population and Communities (SEWPC) for the Natural Resource Management Spatial Information System. The list was produced using the AustralianAustralian Natural Natural Heritage Heritage Assessment Assessment Tool Tool (ANHAT), which analyses data from a range of plant and animal surveys and collections from across Australia to automatically generate a report for each NRM region. Data sources (Appendix 2) include national and state herbaria, museums, state governments, CSIRO, Birds Australia and a range of surveys conducted by or for DEWHA. For each family of plant and animal covered by ANHAT (Appendix 1), this document gives the number of species in the country and how many of them are found in the region. It also identifies species listed as Vulnerable, Critically Endangered, Endangered or Conservation Dependent under the EPBC Act. A biodiversity summary for this region is also available. For more information please see: www.environment.gov.au/heritage/anhat/index.html Limitations • ANHAT currently contains information on the distribution of over 30,000 Australian taxa. This includes all mammals, birds, reptiles, frogs and fish, 137 families of vascular plants (over 15,000 species) and a range of invertebrate groups. Groups notnot yet yet covered covered in inANHAT ANHAT are notnot included included in in the the list. list. • The data used come from authoritative sources, but they are not perfect. All species names have been confirmed as valid species names, but it is not possible to confirm all species locations. -

South American Cacti in Time and Space: Studies on the Diversification of the Tribe Cereeae, with Particular Focus on Subtribe Trichocereinae (Cactaceae)
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2013 South American Cacti in time and space: studies on the diversification of the tribe Cereeae, with particular focus on subtribe Trichocereinae (Cactaceae) Lendel, Anita Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-93287 Dissertation Published Version Originally published at: Lendel, Anita. South American Cacti in time and space: studies on the diversification of the tribe Cereeae, with particular focus on subtribe Trichocereinae (Cactaceae). 2013, University of Zurich, Faculty of Science. South American Cacti in Time and Space: Studies on the Diversification of the Tribe Cereeae, with Particular Focus on Subtribe Trichocereinae (Cactaceae) _________________________________________________________________________________ Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr.sc.nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Anita Lendel aus Kroatien Promotionskomitee: Prof. Dr. H. Peter Linder (Vorsitz) PD. Dr. Reto Nyffeler Prof. Dr. Elena Conti Zürich, 2013 Table of Contents Acknowledgments 1 Introduction 3 Chapter 1. Phylogenetics and taxonomy of the tribe Cereeae s.l., with particular focus 15 on the subtribe Trichocereinae (Cactaceae – Cactoideae) Chapter 2. Floral evolution in the South American tribe Cereeae s.l. (Cactaceae: 53 Cactoideae): Pollination syndromes in a comparative phylogenetic context Chapter 3. Contemporaneous and recent radiations of the world’s major succulent 86 plant lineages Chapter 4. Tackling the molecular dating paradox: underestimated pitfalls and best 121 strategies when fossils are scarce Outlook and Future Research 207 Curriculum Vitae 209 Summary 211 Zusammenfassung 213 Acknowledgments I really believe that no one can go through the process of doing a PhD and come out without being changed at a very profound level. -

AIPC Special Issue 1: the Petiolaris Complex
SPECIAL ISSUE 1 Special Edition Special Issue 1 - English version of AIPCMagazine N° 7 – 2007 The Drosera petiolaris complex Publication reserved to AIPC members. by Maurizio Saroldi Edited byAIPC - Associazione Italiana Piante Carnivore www.aipcnet.it Special Issue N°1 English version of AIPCMagazine N° 7 – 2007 Editorial Index Graziano Fiocca AIPCMagazine is edited by AIPC (Italian Carnivorous Plant Association) 1 Special Edition Reg.Tribunale Venezia n.08 del 5 Maggio 2006 Editor in Chief: Cristina De Rossi The Drosera petiolaris complex Dear readers, by Maurizio Saroldi every year, the AIPC offers to its members 4 issues of its maga- zine, dedicated to several topics on the wonderful world of carni- 6 Introduction vorous plants. 8 Habitat and growth cycle In 2007 we begun an experiment: to devote an entire issue to President one single topic. We asked Maurizio Saroldi, the winner of the Graziella Antonello 16 The Species [email protected] 2006 Italian Carnivorous Olympics, for a personal account of his 50 Cultivation experiences with growing the fascinating petiolaris-complex sun- Secretary 56 Growers, a comparison dews. As a result, in November 2007, the first monograph came Andrea Scaccabarozzi [email protected] to light: AIPCMagazine 7, a double issue comprising 64 pages 59 Where to buy of incredible beauty. The foreign growers who received this work Treasurer 60 Aknowledgements voiced their appreciation, but also their regret at not being able to Luigi Tartaglia read the text, which of course, was in Italian. [email protected] 62 Bibliography Editor For this reason we have now decided to prepare an English Editorial Group version of the monograph.