145053318.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Measuring and Predicting Sooting Tendencies of Oxygenates, Alkanes, Alkenes, Cycloalkanes, and Aromatics on a Unified Scale
Measuring and Predicting Sooting Tendencies of Oxygenates, Alkanes, Alkenes, Cycloalkanes, and Aromatics on a Unified Scale Dhrubajyoti D. Dasa,1, Peter St. Johnb,1, Charles S. McEnallya,∗, Seonah Kimb, Lisa D. Pfefferlea aYale University, Department of Chemical and Environmental Engineering, New Haven CT 06520 bNational Renewable Energy Laboratory, Golden CO 80401 Abstract Soot from internal combustion engines negatively affects health and climate. Soot emissions might be reduced through the expanded usage of appropriate biomass-derived fuels. Databases of sooting indices, based on measuring some aspect of sooting behavior in a standardized combustion environment, are useful in providing information on the comparative sooting tendencies of different fuels or pure compounds. However, newer biofuels have varied chemical structures including both aromatic and oxygenated functional groups, making an accurate measurement or prediction of their sooting tendency difficult. In this work, we propose a unified sooting tendency database for pure compounds, including both regular and oxygenated hydrocarbons, which is based on combining two disparate databases of yield-based sooting tendency measurements in the literature. Unification of the different databases was made possible by leveraging the greater dynamic range of the color ratio pyrometry soot diagnostic. This unified database contains a substantial number of pure compounds (≥ 400 total) from multiple categories of hydrocarbons important in modern fuels and establishes the sooting tendencies of aromatic and oxygenated hydrocarbons on the same numeric scale for the first time. Using this unified sooting tendency database, we have developed a predictive model for sooting behavior applicable to a broad range of hydrocarbons and oxygenated hydrocarbons. The model decomposes each compound into single-carbon fragments and assigns a sooting tendency contribution to each fragment based on regression against the unified database. -

Nicolet Condensed Phase Academic Sampler
Nicolet Condensed Phase Academic Sampler Library Listing – 1,000 spectra This high resolution format library is suited to the needs of academic institutions and small QC labs. Chosen by chemistry professors from many disciplines, it includes spectra of chemicals used in a wide range of common laboratory experiments. The Nicolet Condensed Phase Academic Sampler includes 1,000 spectra of common chemicals representing the major functional groups and combinations of functional groups which are most likely to be observed in academic chemistry laboratories. These chemicals are also important building blocks commonly found in industrial applications. Thermo Nicolet Condensed Phase Academic Sampler Index Compound Name Index Compound Name 353 (+)-2-Phenyl-1-propanol, 97% 164 1,2,4,5-Tetramethylbenzene, 98% 768 (+)-4-Cholesten-3-one 161 1,2,4-Trimethylbenzene, 99+% 290 (+)-a-Lactose 254 1,2-Butanediol, 98% 262 (+)-b-Citronellol, 95% 499 1,2-Diaminopropane, 99% 344 (+/-)-1-Phenyl-1-propanol, 99% 128 1,2-Dibromoethylene, 98%, (Z) + (E) 101 (+/-)-2-Bromopentane, 97% 106 1,2-Dichloroethane, 99+% 228 (+/-)-2-Butanol, 99% 110 1,2-Dichloropropane, 99% 233 (+/-)-2-Heptanol, 96% 258 1,2-Pentanediol, tech., 95% 402 (+/-)-Camphor, 97% 550 1,2-Phenylenediamine, 98% 553 (+/-)-Epinephrine, 99% 77 1,3,5,7-Cyclooctatetraene, 98% 280 (+/-)-Isoborneol, 85% 45 1,3,5-Hexatriene 706 (+/-)-Warfarin, 98% 74 1,3-Cycloheptadiene, 97% 648 (+/-)-sec-Butyl acetate, 99% 72 1,3-Cyclohexadiene, 96% 490 (+/-)-sec-Butylamine, 99% 388 1,3-Cyclohexanedione, 97% 279 (-)-Borneol, -

Synthetic and Mechanistic Investigations in Organophosphorus Chemistry Towards an Alternative Mitsunobu Protocol
Synthetic and Mechanistic Investigations of Some Novel Organophosphorus Reagents Author Fairfull-Smith, Kathryn Elizabeth Published 2004 Thesis Type Thesis (PhD Doctorate) School School of Science DOI https://doi.org/10.25904/1912/3875 Copyright Statement The author owns the copyright in this thesis, unless stated otherwise. Downloaded from http://hdl.handle.net/10072/367534 Griffith Research Online https://research-repository.griffith.edu.au SYNTHETIC AND MECHANISTIC INVESTIGATIONS OF SOME NOVEL ORGANOPHOSPHORUS REAGENTS KATHRYN ELIZABETH FAIRFULL-SMITH (née ELSON) BSc (Hons) School of Science Faculty of Science Griffith University Submitted in fulfilment of the requirements of the Degree of Doctor of Philosophy May 2004 i Statement of Originality This work has not previously been submitted for a degree or diploma in any University. To the best of my knowledge and belief, this thesis contains no material previously published or written by another person except where due reference is made in the thesis itself. Kathryn Fairfull-Smith (née Elson) BSc (Hons) ii Preface Unless otherwise stated, the results in this thesis are those of the author. Parts of this work have appeared elsewhere. Refereed journal publications are submitted with the dissertation and are presented in Appendix Two. Refereed Journal Publications ‘The Hendrickson reagent and the Mitsunobu reaction : a mechanistic study’ Kathryn E. Elson, Ian D. Jenkins and Wendy A. Loughlin, Org. Biomol. Chem., 2003, 1, 2958-2965. ‘Cyclic analogues of the Hendrickson ‘POP’ reagent’ Kathryn E. Elson, Ian D. Jenkins and Wendy A. Loughlin, Aust. J. Chem., 2004, 57, 371-376. ‘Polymer-supported triphenylphosphine ditriflate : a novel dehydrating reagent’ Kathryn E. Elson, Ian D. -
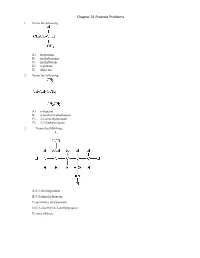
Chapter 21 Practice Problems 1
Chapter 21 Practice Problems 1. Name the following: A) isopropane B) methylpentane C) methylbutane D) n-pentane E) dodecane 2. Name the following: A) n-heptane B) 2-methyl-2-ethylbutane C) 3,3-dimethylpentane D) 2,2-diethylpropane 3. Name the following: A) 2,4-diethylpentane B) 3,5-dimethylheptane C) secondary ethylpentane D) 2,3-dimethyl-2,3-diethylpropane E) none of these 4. In lecture, a professor named a molecule 2-ethyl-4-tert-butylpentane. A student pointed out that the name was incorrect. What is the correct systematic name for the molecule? A) 2-t-butyl-5-methylhexane B) 2-ethyl-4,5,5-trimethylhexane C) 3,5,6,6-tetramethylheptane D) 2,2,3,5-tetramethylheptane E) undecane 5. Structural isomers have A) different molecular formulas and different structures. B) different molecular formulas but the same structure. C) the same molecular formula and the same structure. D) the same molecular formula but different structures. E) none of these 6. How many structural isomers does propane have? A) 3 B) 2 C) 1 D) 5 E) 4 7. The product of ethane undergoing dehydrogenation is called A) propene. B) methene. C) ethene. D) propane. E) none of these 8. Which of the following, upon reacting with oxygen, would form the greatest amount of carbon dioxide? A) n-pentane B) isopentane C) neopentane D) Two of these would form equal amounts. E) All of these would form equal amounts. 9. Which of the following has the lowest boiling point? A) methane B) butane C) ethane D) propane E) All of these have the same boiling point. -

Δ13c of Aromatic Compounds in Sediments, Oils And
1 δ13C of aromatic compounds in sediments, oils and 2 atmospheric emissions: a review 3 Alex I. Holman a, Kliti Grice a* 4 5 a Western Australia Organic and Isotope Geochemistry Centre, The Institute for 6 Geoscience Research, School of Earth and Planetary Sciences, Curtin University, 7 GPO Box U1987, Perth, WA 6845, Australia 8 9 * Corresponding author 10 Alex Holman: Tel.: +61(0) 8 9266 9723. E-mail address: [email protected] 11 Kliti Grice: Tel.: +61(0) 8 9266 2474. E-mail address: [email protected] 12 13 14 15 16 17 18 19 Abstract 20 This review discusses major applications of stable carbon isotopic 21 measurements of aromatic compounds, along with some specific technical aspects 22 including purification of aromatic fractions for baseline separation. δ13C 23 measurements of organic matter (OM) in sediments and oils are routine in all 24 fields of organic geochemistry, but they are predominantly done on saturated 25 compounds. Aromatic compounds are important contributors to sedimentary 26 organic matter, and provide indication of diagenetic processes, OM source, and 27 thermal maturity. Studies have found evidence for a small 13C-enrichment during 28 diagenetic aromatisation of approximately 1 to 2 ‰, but the formation of polycyclic 29 aromatic hydrocarbons (PAHs) from combustion and hydrothermal processes 30 seems to produce no effect. Likewise, maturation and biodegradation also produce 31 only small isotopic effects. An early application of δ13C of aromatic compounds was 32 in the classification of oil families by source. Bulk measurements have had some 33 success in differentiating marine and terrigenous oils, but were not accurate in all 34 settings. -

Effects of Sulfide Minerals on Aromatic Maturity Parameters
Organic Geochemistry 76 (2014) 270–277 Contents lists available at ScienceDirect Organic Geochemistry journal homepage: www.elsevier.com/locate/orggeochem Effects of sulfide minerals on aromatic maturity parameters: Laboratory investigation using micro-scale sealed vessel pyrolysis ⇑ ⇑ Alex I. Holman a, , Paul F. Greenwood a,b,c, Jochen J. Brocks d, Kliti Grice a, a Western Australia Organic and Isotope Geochemistry Centre, Department of Chemistry, The Institute for Geoscience Research, Curtin University, GPO Box U1987, Perth, WA 6845, Australia b Centre for Exploration Targeting, University of Western Australia, Crawley, WA 6009, Australia c WA Biogeochemistry Centre, University of Western Australia, Crawley, WA 6009, Australia d Research School of Earth Sciences, The Australian National University, Canberra, ACT 0200, Australia article info abstract Article history: Sedimentary organic matter from the Here’s Your Chance (HYC) Pb–Zn–Ag deposit (McArthur Basin, Received 19 December 2013 Northern Territory, Australia) displays increased thermal maturity compared to nearby non-mineralised Received in revised form 28 August 2014 sediments. Micro-scale sealed vessel pyrolysis (MSSVpy) of an immature, organic rich sediment from the Accepted 2 September 2014 host Barney Creek Formation (BCF) was used to simulate the thermal maturation of OM from the HYC Available online 16 September 2014 deposit, and to assess the effect of sulfide minerals on organic maturation processes. MSSVpy at increas- ing temperatures (300, 330 and 360 °C) resulted in increased methylphenanthrene maturity ratios which Keywords: were within the range reported for bitumen extracted from HYC sediments. The methylphenanthrene Micro-scale sealed vessel pyrolysis index ratio from MSSVpy of the BCF sample was lower than in HYC, due to a reduced proportion of meth- Polycyclic aromatic hydrocarbon Thermal maturity ylated phenanthrenes. -

Thesis Has Been Carried out in the School of Pharmacy and Pharmacology and in the School of Biology and Biochemistry, Under the Supervision of Dr Michael D
University of Bath PHD Inhibitors of DNA repair processes as potentiating drugs in cancer radiotherapy and chemotherapy Watson, Corrine Yvonne Award date: 1997 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 10. Oct. 2021 Inhibitors of DNA Repair Processes as Potentiating Drugs in Cancer Radiotherapy and Chemotherapy submitted by Corrine Yvonne Watson for the degree of PhD of the University of Bath 1997 The research work in this thesis has been carried out in the School of Pharmacy and Pharmacology and in the School of Biology and Biochemistry, under the supervision of Dr Michael D. Threadgill and Dr William J. D. Whish. COPYRIGHT Attention is drawn to the fact that copyright of this thesis rests with its author. -

Organic Chemistry/Fourth Edition: E-Text
CHAPTER 17 ALDEHYDES AND KETONES: NUCLEOPHILIC ADDITION TO THE CARBONYL GROUP O X ldehydes and ketones contain an acyl group RC± bonded either to hydrogen or Ato another carbon. O O O X X X HCH RCH RCRЈ Formaldehyde Aldehyde Ketone Although the present chapter includes the usual collection of topics designed to acquaint us with a particular class of compounds, its central theme is a fundamental reaction type, nucleophilic addition to carbonyl groups. The principles of nucleophilic addition to alde- hydes and ketones developed here will be seen to have broad applicability in later chap- ters when transformations of various derivatives of carboxylic acids are discussed. 17.1 NOMENCLATURE O X The longest continuous chain that contains the ±CH group provides the base name for aldehydes. The -e ending of the corresponding alkane name is replaced by -al, and sub- stituents are specified in the usual way. It is not necessary to specify the location of O X the ±CH group in the name, since the chain must be numbered by starting with this group as C-1. The suffix -dial is added to the appropriate alkane name when the com- pound contains two aldehyde functions.* * The -e ending of an alkane name is dropped before a suffix beginning with a vowel (-al) and retained be- fore one beginning with a consonant (-dial). 654 Back Forward Main Menu TOC Study Guide TOC Student OLC MHHE Website 17.1 Nomenclature 655 CH3 O O O O CH3CCH2CH2CH CH2 CHCH2CH2CH2CH HCCHCH CH3 4,4-Dimethylpentanal 5-Hexenal 2-Phenylpropanedial When a formyl group (±CHœO) is attached to a ring, the ring name is followed by the suffix -carbaldehyde. -

ELECTRONICS AUTHENTICITY TESTING USING COMPREHENSIVE TWO-DIMENSIONAL GAS CHROMATOGRAPHY by Joseph C
ELECTRONICS AUTHENTICITY TESTING USING COMPREHENSIVE TWO-DIMENSIONAL GAS CHROMATOGRAPHY by Joseph C. Cacciatore A Thesis Submitted to the Faculty of Purdue University In Partial Fulfillment of the Requirements for the degree of Master of Science School of Engineering Technology West Lafayette, Indiana December 2019 2 THE PURDUE UNIVERSITY GRADUATE SCHOOL STATEMENT OF COMMITTEE APPROVAL Dr. J. Eric Dietz, Chair Department of Computer and Information Technology Dr. Gozdem Kilaz, Chair School of Engineering Technology Dr. Ken Burbank School of Engineering Technology Approved by: Dr. Duane Dunlap Head of the Graduate Program 2 TABLE OF CONTENTS LIST OF TABLES .......................................................................................................................... 5 LIST OF FIGURES ........................................................................................................................ 6 LIST OF ABBREVIATIONS ......................................................................................................... 7 DEFINITIONS ................................................................................................................................ 8 ABSTRACT .................................................................................................................................... 9 INTRODUCTION .............................................................................................. 10 1.1 Introduction to the Problem ............................................................................................. -

1,2,4-Trimethylbenzene Transformation Reaction Compared with Its Transalkylation Reaction with Toluene Over USY-Zeolite Catalyst
1,2,4-Trimethylbenzene Transformation Reaction Compared with its Transalkylation Reaction with Toluene over USY-Zeolite Catalyst Sulaiman Al-Khattaf*, Nasir M. Tukur, and Adnan Al-Amer Chemical Engineering Department, King Fahd University of Petroleum & Minerals Dhahran 31261, Saudi Arabia Abstract 1,2,4-Trimethyl benzene (TMB) transalkylation with toluene has been studied over USY-zeolite type catalyst using a riser simulator that mimics the operation of a fluidized-bed reactor. 50:50 wt% reaction mixtures of TMB and toluene were used for the transalkylation reaction. The range of temperature investigated was 400-500 oC and time on stream ranging from 3 to 15 seconds. The effect of reaction conditions on the variation of p-xylene to o-xylene products ratio (P/O), distribution of trimethylbenzene (TMB) isomers (1,3,5-to-1,2,3-) and values of xylene/tetramethylbenzenes (X/TeMB) ratios are reported. Comparisons are made between the results of the transalkylation reaction with the results of pure 1,2,4-TMB and toluene reactions earlier reported. Toluene that was found almost inactive, became reactive upon blending with 1,2,4-TMB. This shows that toluene would rather accept a methyl group to transform to xylene than to loose a methyl group to form benzene under the present experimental condition with pressures around ambient. The experimental results were modeled using quasi-steady state approximation. Kinetic parameters for the 1,2,4-TMB disappearance during the transalkylation reaction, and in its conversion into isomerization and disproportionation products were calculated using the catalyst activity decay function based on time on stream (TOS). -

Journal Name Dynamic Article Links ►
Journal Name Dynamic Article Links ► Cite this: DOI: 10.1039/c0xx00000x www.rsc.org/xxxxxx ARTICLE TYPE A Convenient & Mild Chromatography-Free Method for the Purification of the Products of Wittig and Appel Reactions† Peter A. Byrne,a Kamalraj V. Rajendrana and Declan G. Gilheany*a 5 Received (in XXX, XXX) Xth XXXXXXXXX 20XX, Accepted Xth XXXXXXXXX 20XX DOI: 10.1039/b000000x A mild method for the facile removal of phosphine oxide from On a laboratory scale, phosphine oxide removal generally the crude products of Wittig and Appel reactions is described. necessitates the use of column chromatography. This presents Work-up with oxalyl chloride to generate insoluble 50 problems if the product is sensitive to decomposition or 10 chlorophosphonium salt (CPS) yields phosphorus-free isomerisation through contact with the stationary phase or by products for a wide variety of these reactions. The CPS light, or if it is sensitive to decomposition in air. For example, product can be further converted into phosphine. certain alkenes produced in Wittig reactions have been shown to be sensitive to isomerisation by light, or by contact with silica or 12 Phosphines and reagents derived from them are ubiquitous 55 alumina. Thus, there is a strong imperative for the development reagents in organic chemistry used, for example, in the Wittig of chromatography-free phosphine oxide removal. Reported 1 2 3 15 reaction, in Appel and Mitsunobu conditions and in the aza- methods addressing this need include modification of Wittig reaction.4 However the phosphine oxide by-product of phosphonium ylides to furnish a phosphine oxide by-product that these reactions poses significant problems, both of separation and may be removed by aqueous work-up13,14 and the use of water- disposal, leading to poor atom efficiency 5,6,7 and requiring 60 soluble and hydrolytically stable ylides to allow reactions to be complex procedures,7,8 particularly for large-scale reactions. -

Biosynthesis of C11 Hydrocarbons in the Brown Alga Ectocarpus
Biosynthesis of C11 hydrocarbons in the brown alga Ectocarpus siliculosus Dissertation Zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat) Vorgelegt dem Rat der Chemisch-Geowissenschafltlichen Fakultät der Friedrich-Schiller- Universität Jena von Diplom-Chemiker Fabio Rui geboren am 04.02.1975 in Pordenone Gutachter: 1. Prof. Dr. Wilhelm Boland Department of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Jena 2. Prof. Dr. Reiner Beckert Institute für Organische Chemie und Makromolekulare Chemie, Friedrich Schiller Universität, Jena Tag der öffentlicher Verteidigung: 20.05.2009 List of abbreviations CLS closed loop stripping DCC dicyclohexylcarbodiimide DiHODA dihydroxyoctadecadienoic acid DMAP 4-Dimethylaminopyridine DMSO dimethylsulfoxide GC/MS gas chromatography/mass spectrometry HETE hydroxyeicosatetraenoic acid HKR hydrolytic kinetic resolution HPETE hydroperoxyeicosatetraenoic acid HPL hydropeoxyde lyase HPOT hydroperoxyoctadecatetraenoic IBX 1-Hydroxy-1,2-benziodoxol-3-(1H)-one 1-Oxide LDS linoleate diol synthase LOX lipoxygenase PFBHA pentafluorobenzyl hydroxylamine PGG2 prostaglandin G2 PGHS prostaglandin H synthase PUFA polyunsaturated fatty acid SPME solid phase microextraction THF tetrahydrofurane Table of contents Biosynthesis of C11 hydrocarbons in the brown alga E. siliculosus List of abbreviations 5 1 Introduction 9 1.1 Chemical ecology of brown algal pheromones 9 1.2 Biosynthesis of C11 hydrocarbons in Ectocarpus siliculosus and other organisms 12 1.3 Selected enzymes catalysing