Δ13c of Aromatic Compounds in Sediments, Oils And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Measuring and Predicting Sooting Tendencies of Oxygenates, Alkanes, Alkenes, Cycloalkanes, and Aromatics on a Unified Scale
Measuring and Predicting Sooting Tendencies of Oxygenates, Alkanes, Alkenes, Cycloalkanes, and Aromatics on a Unified Scale Dhrubajyoti D. Dasa,1, Peter St. Johnb,1, Charles S. McEnallya,∗, Seonah Kimb, Lisa D. Pfefferlea aYale University, Department of Chemical and Environmental Engineering, New Haven CT 06520 bNational Renewable Energy Laboratory, Golden CO 80401 Abstract Soot from internal combustion engines negatively affects health and climate. Soot emissions might be reduced through the expanded usage of appropriate biomass-derived fuels. Databases of sooting indices, based on measuring some aspect of sooting behavior in a standardized combustion environment, are useful in providing information on the comparative sooting tendencies of different fuels or pure compounds. However, newer biofuels have varied chemical structures including both aromatic and oxygenated functional groups, making an accurate measurement or prediction of their sooting tendency difficult. In this work, we propose a unified sooting tendency database for pure compounds, including both regular and oxygenated hydrocarbons, which is based on combining two disparate databases of yield-based sooting tendency measurements in the literature. Unification of the different databases was made possible by leveraging the greater dynamic range of the color ratio pyrometry soot diagnostic. This unified database contains a substantial number of pure compounds (≥ 400 total) from multiple categories of hydrocarbons important in modern fuels and establishes the sooting tendencies of aromatic and oxygenated hydrocarbons on the same numeric scale for the first time. Using this unified sooting tendency database, we have developed a predictive model for sooting behavior applicable to a broad range of hydrocarbons and oxygenated hydrocarbons. The model decomposes each compound into single-carbon fragments and assigns a sooting tendency contribution to each fragment based on regression against the unified database. -
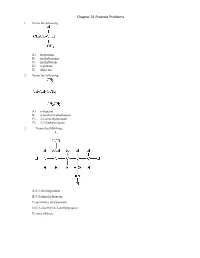
Chapter 21 Practice Problems 1
Chapter 21 Practice Problems 1. Name the following: A) isopropane B) methylpentane C) methylbutane D) n-pentane E) dodecane 2. Name the following: A) n-heptane B) 2-methyl-2-ethylbutane C) 3,3-dimethylpentane D) 2,2-diethylpropane 3. Name the following: A) 2,4-diethylpentane B) 3,5-dimethylheptane C) secondary ethylpentane D) 2,3-dimethyl-2,3-diethylpropane E) none of these 4. In lecture, a professor named a molecule 2-ethyl-4-tert-butylpentane. A student pointed out that the name was incorrect. What is the correct systematic name for the molecule? A) 2-t-butyl-5-methylhexane B) 2-ethyl-4,5,5-trimethylhexane C) 3,5,6,6-tetramethylheptane D) 2,2,3,5-tetramethylheptane E) undecane 5. Structural isomers have A) different molecular formulas and different structures. B) different molecular formulas but the same structure. C) the same molecular formula and the same structure. D) the same molecular formula but different structures. E) none of these 6. How many structural isomers does propane have? A) 3 B) 2 C) 1 D) 5 E) 4 7. The product of ethane undergoing dehydrogenation is called A) propene. B) methene. C) ethene. D) propane. E) none of these 8. Which of the following, upon reacting with oxygen, would form the greatest amount of carbon dioxide? A) n-pentane B) isopentane C) neopentane D) Two of these would form equal amounts. E) All of these would form equal amounts. 9. Which of the following has the lowest boiling point? A) methane B) butane C) ethane D) propane E) All of these have the same boiling point. -

Effects of Sulfide Minerals on Aromatic Maturity Parameters
Organic Geochemistry 76 (2014) 270–277 Contents lists available at ScienceDirect Organic Geochemistry journal homepage: www.elsevier.com/locate/orggeochem Effects of sulfide minerals on aromatic maturity parameters: Laboratory investigation using micro-scale sealed vessel pyrolysis ⇑ ⇑ Alex I. Holman a, , Paul F. Greenwood a,b,c, Jochen J. Brocks d, Kliti Grice a, a Western Australia Organic and Isotope Geochemistry Centre, Department of Chemistry, The Institute for Geoscience Research, Curtin University, GPO Box U1987, Perth, WA 6845, Australia b Centre for Exploration Targeting, University of Western Australia, Crawley, WA 6009, Australia c WA Biogeochemistry Centre, University of Western Australia, Crawley, WA 6009, Australia d Research School of Earth Sciences, The Australian National University, Canberra, ACT 0200, Australia article info abstract Article history: Sedimentary organic matter from the Here’s Your Chance (HYC) Pb–Zn–Ag deposit (McArthur Basin, Received 19 December 2013 Northern Territory, Australia) displays increased thermal maturity compared to nearby non-mineralised Received in revised form 28 August 2014 sediments. Micro-scale sealed vessel pyrolysis (MSSVpy) of an immature, organic rich sediment from the Accepted 2 September 2014 host Barney Creek Formation (BCF) was used to simulate the thermal maturation of OM from the HYC Available online 16 September 2014 deposit, and to assess the effect of sulfide minerals on organic maturation processes. MSSVpy at increas- ing temperatures (300, 330 and 360 °C) resulted in increased methylphenanthrene maturity ratios which Keywords: were within the range reported for bitumen extracted from HYC sediments. The methylphenanthrene Micro-scale sealed vessel pyrolysis index ratio from MSSVpy of the BCF sample was lower than in HYC, due to a reduced proportion of meth- Polycyclic aromatic hydrocarbon Thermal maturity ylated phenanthrenes. -

Featured: Simon Brassell 9/18/2020 PRE-SCRIPT in the Fourth Episode
Featured: Simon Brassell 9/18/2020 PRE-SCRIPT In the fourth episode of BIOmarkers, the audio series that archives the oral history of organic geochemistry, we speak with Dr. Simon Brassell, Professor of Geological Sciences, Biogeochemistry, and Molecular Organic Geochemistry at the Department of Earth and Atmospheric Sciences at Indiana University Bloomington. In his interview, Simon discusses his time at the University of Bristol, proxies, and what piques his interest today. SCRIPT Fatima Husain: Welcome to BIOmarkers, an audio series that archives the oral history of organic geochemistry. I’m your host, Fatima Husain, and I’m here today with my series co-creators and fellow organic geochemists Angel Mojarro and Juliana Drozd. Angel Mojarro: For today’s episode, we spoke with Dr. Simon Brassell. Simon Brassell: I'm Simon Brassell, I'm a professor of Earth and Planetary Sciences at Indiana University. When anyone asked me what I am, I would say I'm a molecular organic geochemistry and molecular bio bio geochemist, because my area of expertise started in looking at biomarkers, and I have pretty much stayed in that area all along. Juliana Drozd: We spoke with Simon briefly in between sessions at the 2019 International Meeting on Organic Geochemistry about his career pathway and insights into the field. Fatima Husain: It turns out, this isn’t too hard to do: Simon Brassell: This is about something like the 14th, 15th time I've come to IMOG and every year there are two, two. There's one meeting each year that I always try and get to and it's the IMOG and then even years, it's the Gordon research conference on organic chemistry because I learn more at those meetings about the specifics of the subject than I do at any other bigger or meetings that aren't a less focused on organic geochemistry, and it's always great to come back and see people who I've known some for 10 years, some for 20 years, some 30 years, some 40 years. -

1,2,4-Trimethylbenzene Transformation Reaction Compared with Its Transalkylation Reaction with Toluene Over USY-Zeolite Catalyst
1,2,4-Trimethylbenzene Transformation Reaction Compared with its Transalkylation Reaction with Toluene over USY-Zeolite Catalyst Sulaiman Al-Khattaf*, Nasir M. Tukur, and Adnan Al-Amer Chemical Engineering Department, King Fahd University of Petroleum & Minerals Dhahran 31261, Saudi Arabia Abstract 1,2,4-Trimethyl benzene (TMB) transalkylation with toluene has been studied over USY-zeolite type catalyst using a riser simulator that mimics the operation of a fluidized-bed reactor. 50:50 wt% reaction mixtures of TMB and toluene were used for the transalkylation reaction. The range of temperature investigated was 400-500 oC and time on stream ranging from 3 to 15 seconds. The effect of reaction conditions on the variation of p-xylene to o-xylene products ratio (P/O), distribution of trimethylbenzene (TMB) isomers (1,3,5-to-1,2,3-) and values of xylene/tetramethylbenzenes (X/TeMB) ratios are reported. Comparisons are made between the results of the transalkylation reaction with the results of pure 1,2,4-TMB and toluene reactions earlier reported. Toluene that was found almost inactive, became reactive upon blending with 1,2,4-TMB. This shows that toluene would rather accept a methyl group to transform to xylene than to loose a methyl group to form benzene under the present experimental condition with pressures around ambient. The experimental results were modeled using quasi-steady state approximation. Kinetic parameters for the 1,2,4-TMB disappearance during the transalkylation reaction, and in its conversion into isomerization and disproportionation products were calculated using the catalyst activity decay function based on time on stream (TOS). -

This Article Was Published in an Elsevier Journal. the Attached Copy
This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author’s institution, sharing with colleagues and providing to institution administration. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright Author's personal copy Available online at www.sciencedirect.com Geochimica et Cosmochimica Acta 72 (2008) 1396–1414 www.elsevier.com/locate/gca Okenane, a biomarker for purple sulfur bacteria (Chromatiaceae), and other new carotenoid derivatives from the 1640 Ma Barney Creek Formation Jochen J. Brocks a,*, Philippe Schaeffer b a Research School of Earth Sciences and Centre for Macroevolution and Macroecology, The Australian National University, Canberra, ACT 0200, Australia b Laboratoire de Ge´ochimie Bio-organique, CNRS UMR 7177, Ecole Europe´enne de Chimie, Polyme`res et Mate´riaux, 25 rue Becquerel, 67200 Strasbourg, France Received 20 June 2007; accepted in revised form 12 December 2007; available online 23 December 2007 Abstract Carbonates of the 1640 million years (Ma) old Barney Creek Formation (BCF), McArthur Basin, Australia, contain more than 22 different C40 carotenoid derivatives including lycopane, c-carotane, b-carotane, chlorobactane, isorenieratane, b-iso- renieratane, renieratane, b-renierapurpurane, renierapurpurane and the monoaromatic carotenoid okenane. -
![United States Patent [19] [11] Patent Number: 6,147,270 Pazzucconi Et Al](https://docslib.b-cdn.net/cover/4104/united-states-patent-19-11-patent-number-6-147-270-pazzucconi-et-al-1394104.webp)
United States Patent [19] [11] Patent Number: 6,147,270 Pazzucconi Et Al
US006147270A United States Patent [19] [11] Patent Number: 6,147,270 Pazzucconi et al. [45] Date of Patent: Nov. 14, 2000 [54] PROCESS FOR THE PREPARATION OF 5,670,704 9/1997 Hagen et al. ......................... .. 585/471 2,6-DIMETHYLNAPHTHALENE USING A 5,672,799 9/1997 Perego et al. ......................... .. 585/467 MTW ZEOLITIC CATALYST FOREIGN PATENT DOCUMENTS [75] Inventors: Giannino Pazzucconi, Broni; Carlo 2 246 788 2/1992 United Kingdom . Perego, Carnate; Roberto Millini, WO 90/03960 4/1990 WIPO. Cerro al Lambro; Francesco Frigerio, OTHER PUBLICATIONS Torre d’lsola; Riccardo Mansani, Sassari; Daniele Rancati, Porto Torres, “MTW”; internet search record, Dec. 1999. all of Italy Primary Examiner—Marian C. Knode [73] Assignee: Enichem S.p.A., S. Donato Milanese, Assistant Examiner—Thuan D. Dang Italy Attorney, Agent, or Firm—Oblon, Spivak, McClelland, Maier & Neustadt, PC. [21] Appl. No.: 09/281,961 [57] ABSTRACT [22] Filed: Mar. 31, 1999 A highly selective process is described for preparing 2,6 dimethylnaphthalene Which comprises reacting a naphtha [30] Foreign Application Priority Data lene hydrocarbon selected from naphthalene, Apr. 17, 1998 [IT] Italy ............................... .. MI98A0809 methylnaphthalenes, dimethylnaphthalenes, trimethylnaphthalenes, polymethylnaphthalenes or their [51] Int. Cl.7 ................................................... .. C07C 15/12 mixtures With one or more benzene hydrocarbons selected [52] US. Cl. ........................................... .. 585/475; 585/471 from benzene, toluene, Xylenes, trimethylbenZenes, [58] Field of Search ................................... .. 585/475, 471, tetramethylbenZenes, pentamethylbenZene and/or 585/470 heXamethylbenZene, under at least partially liquid phase conditions, in the presence of a Zeolite belonging to the [56] References Cited structural type MTW and optionally in the presence of a U.S. -

Disproportionation of 1,2,4-Trimethylbenzene Over Zeolite NU-87
Korean J. Chem. Eng., 17(2), 198-204 (2000) Disproportionation of 1,2,4-Trimethylbenzene over Zeolite NU-87 Se-Ho Park, Jong-Hyung Lee and Hyun-Ku Rhee† School of Chemical Engineering and Institute of Chemical Processes, Seoul National University, Kwanak-ku, Seoul 151-742, Korea (Received 27 September 1999 • accepted 30 December 1999) Abstract−The catalytic properties of zeolite NU-87 were investigated with respect to the disproportionation of 1, 2,4-trimethylbenzene and the results were compared to those obtained over H-beta and H-mordenite with 12-mem- bered ring channel system. In the conversion of 1,2,4-trimethylbenzene, the disproportionation to xylene and tetra- methylbenzene is preferred to the isomerization into 1,2,3- and 1,3,5-isomers over all the three zeolites, but this trend is much more pronounced over HNU-87 owing to its peculiar pore structure. Disproportionation reaction is found to proceed within the micropores of HNU-87, whereas isomerization occurs mainly on the external surface. The high selectivity to disproportionation gives more xylenes and tetramethylbenzenes over HNU-87. The detailed descrip- tions for the product distribution are also reported. Key words: NU-87, 1,2,4-Trimethylbenzene, Disproportionation, Isomerization INTRODUCTION EXPERIMENTAL Disproportionation of trimethylbenzene (TMB) to xylene and 1. Catalysts Preparation = tetramethylbenzene (TeMB) is an important process for the indus- H-mordenite (Engelhard, SiO2/Al2O3 45) and H-beta (PQ Corp., = try, mainly due to the increasing demand for p-xylene to be used SiO2/Al2O3 22) used in this study were taken from commercial for the production of polyester resins. -

Trimethylbenzoic Acids As Metabolite Signatures in the Biogeochemical Evolution of an Aquifer Contaminated with Jet Fuel Hydrocarbons
Journal of Contaminant Hydrology 67 (2003) 177–194 www.elsevier.com/locate/jconhyd Trimethylbenzoic acids as metabolite signatures in the biogeochemical evolution of an aquifer contaminated with jet fuel hydrocarbons J.A. Namocatcata,1, J. Fanga,*, M.J. Barcelonaa,2, A.T.O. Quibuyenb, T.A. Abrajano Jr.c a National Center for Integrated Bioremediation Research and Development, Department of Civil and Environmental Engineering, The University of Michigan, Ann Arbor, MI 48109, USA b Institute of Chemistry, University of the Philippines, Diliman, Quezon City 1101, Philippines c Department of Earth and Environmental Sciences, Rensselaer Polytechnic Institute, Troy, NY 12180, USA Received 24 May 2002; accepted 21 March 2003 Abstract Evolution of trimethylbenzoic acids in the KC-135 aquifer at the former Wurtsmith Air Force Base (WAFB), Oscoda, MI was examined to determine the functionality of trimethylbenzoic acids as key metabolite signatures in the biogeochemical evolution of an aquifer contaminated with JP-4 fuel hydrocarbons. Changes in the composition of trimethylbenzoic acids and the distribution and concentration profiles exhibited by 2,4,6- and 2,3,5-trimethylbenzoic acids temporally and between multilevel wells reflect processes indicative of an actively evolving contaminant plume. The concentration levels of trimethylbenzoic acids were 3–10 orders higher than their tetramethylben- zene precursors, a condition attributed to slow metabolite turnover under sulfidogenic conditions. The observed degradation of tetramethylbenzenes into trimethylbenzoic acids obviates the use of these alkylbenzenes as non-labile tracers for other degradable aromatic hydrocarbons, but provides rare field evidence on the range of high molecular weight alkylbenzenes and isomeric assemblages amenable to anaerobic degradation in situ. -

THE GEOCHEMICAL NEWS Newsletter of the Geochemical Society in Cooperation with the European Association of Geochemistry
January 2005 1 THE GEOCHEMICAL NEWS Newsletter of The Geochemical Society in cooperation with The European Association of Geochemistry In This Issue... An Interview with David Des Marais Geochemistry at Oak Ridge National Laboratory January 2005 Number 122 Newsletter of the Geochemical Society ISSN 0016-7010 2 The Geochemical News EAG OFFICERS - 2005 PRESIDENT Terry Seward, ETH, Zurich PRESIDENT ELECT Bruce Yardley, Leeds, UK OUTGOING PRESIDENT Francis Albarede, Lyon, France TREASURER Catherine Chauvel, Grenoble, France SECRETARY Mark Hodson, Reading, UK EAG COMMITTEE THE GEOCHEMICAL SOCIETY MIRA BAR-MATTHEWS, ISREAL Larryn Diamond, Switzerland Jérôme GAILLARDET, FRANCE Alex Halliday, Switzerland SUSAN STIPP, DENMARK Riccardo Vannucci, Italy The Geochemical Society is a nonprofit scientific society founded to en- GERHARD WORNER, GERMANY Bruce Yardley, UK courage the application of chemistry to the solution of geological and cosmologi- cal problems. Membership is international and diverse in background, encom- passing such fields as organic geochemistry, high- and low-temperature geochem- THE GEOCHEMICAL NEWS istry, petrology, meteoritics, fluid-rock interaction, and isotope geochemistry. The Society produces a Special Publications Series, The Geochemical News (this January 2005 quarterly newsletter), the Reviews in Mineralogy and Geochemistry Series (jointly with the Mineralogical Society of America), the journal Geochimica et Editors Cosmochimica Acta (jointly with the Meteoritical Society), and co-publishes the Johnson R. Haas and Carla M. Koretsky electronic journal G3 (jointly with the American Geophysical Union: AGU); grants Department of Geosciences the V.M. Goldschmidt, F.W. Clarke and Clair C. Patterson Awards, and, jointly Western Michigan University with the European Association of Geochemistry (EAG), the Geochemistry Fel- Kalamazoo, MI 49008 lows title; sponsors the V.M. -

Vascular Plant Biomarker Distributions and Stable Carbon Isotopic
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by NERC Open Research Archive 1 Palaeogeography, Palaeoclimatology, Palaeoecology 2 3 Vascular plant biomarker distributions and stable carbon isotopic 4 signatures from the Middle and Upper Jurassic (Callovian–Kimmeridgian) 5 strata of Staffin Bay, Isle of Skye, northwest Scotland 6 7 Kliti Grice 1,*, Clinton B. Foster 2, James B. Riding 3, Sebastian Naeher 1, Paul F. 8 Greenwood 1,4 9 10 1 WA Organic and Isotope Geochemistry Centre, The Institute for Geoscience Research, 11 Department of Chemistry, Curtin University, GPO Box U1987 Perth, WA 6845, Australia 12 2 Geoscience Australia, GPO Box 378, Canberra, ACT, 2601, Australia 13 3 British Geological Survey, Environmental Science Centre, Keyworth, Nottingham NG12 14 5GG, United Kingdom 15 4 Centre for Exploration Targeting and WA Biogeochemistry Centre (M090), The University 16 of Western Australia, 35 Stirling Highway, Crawley, WA, 6009, Australia 17 18 19 20 21 22 23 * Corresponding author. Tel.: +61 8 9266 2474; Fax: +61 8 9266 2300 24 E-mail address: [email protected] (K. Grice) 1 25 Abstract 26 The molecular and stable carbon isotopic composition of higher plant biomarkers was 27 investigated in Middle to Upper Jurassic strata of the Isle of Skye, northwest Scotland. 28 Aromatic hydrocarbons diagnostic of vascular plants were detected in each of nineteen 29 sedimentary rock samples from the Early Callovian to Early Kimmeridgian interval, a 30 succession rich in fossil fauna including ammonites that define its constituent chronozones. 31 The higher plant parameter (HPP) and higher plant fingerprint (HPF) calculated from the 32 relative abundance of retene, cadalene and 6-isopropyl-1-isohexyl-2-methylnaphthalene (ip- 33 iHMN) exhibit several large fluctuations throughout the Skye succession studied. -

"Hydrocarbons," In: Ullmann's Encyclopedia of Industrial Chemistry
Article No : a13_227 Hydrocarbons KARL GRIESBAUM, Universit€at Karlsruhe (TH), Karlsruhe, Federal Republic of Germany ARNO BEHR, Henkel KGaA, Dusseldorf,€ Federal Republic of Germany DIETER BIEDENKAPP, BASF Aktiengesellschaft, Ludwigshafen, Federal Republic of Germany HEINZ-WERNER VOGES, Huls€ Aktiengesellschaft, Marl, Federal Republic of Germany DOROTHEA GARBE, Haarmann & Reimer GmbH, Holzminden, Federal Republic of Germany CHRISTIAN PAETZ, Bayer AG, Leverkusen, Federal Republic of Germany GERD COLLIN, Ruttgerswerke€ AG, Duisburg, Federal Republic of Germany DIETER MAYER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany HARTMUT Ho€KE, Ruttgerswerke€ AG, Castrop-Rauxel, Federal Republic of Germany 1. Saturated Hydrocarbons ............ 134 3.7. Cumene ......................... 163 1.1. Physical Properties ................ 134 3.8. Diisopropylbenzenes ............... 164 1.2. Chemical Properties ............... 134 3.9. Cymenes; C4- and C5-Alkylaromatic 1.3. Production ....................... 134 Compounds ...................... 165 1.3.1. From Natural Gas and Petroleum . .... 135 3.10. Monoalkylbenzenes with Alkyl Groups 1.3.2. From Coal and Coal-Derived Products . 138 >C10 ........................... 166 1.3.3. By Synthesis and by Conversion of other 3.11. Diphenylmethane .................. 167 Hydrocarbons . .................. 139 4. Biphenyls and Polyphenyls .......... 168 1.4. Uses ............................ 140 4.1. Biphenyl......................... 168 1.5. Individual Saturated Hydrocarbons ... 142 4.2. Terphenyls......................