University of Florida Thesis Or Dissertation Formatting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

3-D Nano and Micro Structures
TM Vol. 3, No. 1 3-D Nano and Micro Structures Inks for Direct-Write 3-D Assembly Colloidal Crystal Templating Small Structures Inspiring Electrospinning Big Technologies Quantum Dots: Nanoscale Synthesis and Micron-Scale Applications 2 Introduction TM Welcome to the first 2008 issue of Material Matters™, focusing on 3-dimensional (3D) micro- and nanostructures. New techniques to create materials ordered at micro- Vol. 3 No. 1 and nanoscale drive scientific and technological advances in many areas of science and engineering. For example, 3D-patterned metal oxides could enable construction of micro-fuel cells and high-capacity batteries smaller and more energy efficient compared to presently available devices. Ordered porous materials with exceedingly large surface areas are good candidates for highly efficient catalysts and sensors. Aldrich Chemical Co., Inc. New methods for periodic patterning of semiconductor materials are fundamental Sigma-Aldrich Corporation to next-generation electronics, while individual nanoscale semiconductor structures, 6000 N. Teutonia Ave. known as quantum dots (QDs), have remarkable optical properties for optoelectronics Milwaukee, WI 53209, USA and imaging applications. And biomedical engineers are using tailored nanofibers and patterned polymeric structures to develop tissue-engineering scaffolds, drug-delivery devices and microfluidic networks. To Place Orders The broad diversity of potentially relevant material types and architectures underscores Telephone 800-325-3010 (USA) the need for new approaches to make 3D micro- and nanostructures. In this issue, FAX 800-325-5052 (USA) Introduction Professor Jennifer Lewis (University of Illinois at Urbana-Champaign) describes direct- write printing of 3D periodic arrays. This work depends on formulation of suitable Customer & Technical Services inks, many of which can be prepared using particles and polyelectrolytes available Customer Inquiries 800-325-3010 from Sigma-Aldrich®. -
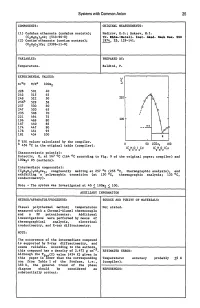
Systems with Common Anion 25
Systems with Common Anion 25 COMPONENTS: ORIGINAL MEASUREMENTS: (1) Cadmium ethanoate (cadmium acetate); Nadirov, E.G.; Bakeev, M.I. (C2H302)2Cd; [543-90-8] Tr. KhilR.-Ketall. IDst. Akad. Nauk Kaz. 8SR (2) Cesium ethanoate (cesium acetate); 1974, 25, 129-141. (C2H302)Cs; [3396-11-0] VARIABLES: PREPARED BY: Temperature. Baldini, P. EXPERIMENTAL VALUES: 228 501 40 242 515 45 249 522 50 250 256b 529 58 257 530 60 ,f 247 520 65 , 235 508 70 , 221 494 75 196 469 80 200 167 440 85 __ ..!!.2. 174 447 90 178 451 95 181 454 100 E a T/K values· calculated by the compiler. 50 lOOx 100 b 456 °c in the original table (compiler). 2 (C H 0 1Cs 2 3 2 Characteristic point(s): Eutectic, E, at 167 °c (164 °c according to Fig. 9 of the original paper; compiler) and 10Ox2= 85 (authors). Intermediate compound(s): (C2H302)7Cd2Cs3' congruently melting at 257 °c (255 °c, thermographic analysis), and exhibiting a polymorphic transition (at 130 °c, thermographic analysis; 133 °C, conductometry). Note - The system was investigated at 401~ lO0x2 ~ 100. AUXILIARY INFORMATION METHOD/APPARATUS/PROCEDURE: SOURCE AND PURITY OF MATERIALS: Visual polythermal method; temperatures Not stated. measured with a Chromel-Alumel thermocouple and a PP potentiometer. Additional investigations were performed by means of thermographical analysis, electrical conductometry, and X-ray diffractometry. NOTE: The occurrence of the intermediate compound is supported by X-ray diffractometry, and seems reliable. According to the authors~ this compound has a density of 2.472 g cm- • ESTIMATED ERROR: Although the Tfus(2) value (454 K) given in this paper is lower than the corresponding Temperature: accuracy probably +2 K one from Table 1 of the Preface, i.e., (compiler) • 463 K, the general trend of the phase diagram should be considered as REFERENCES: substantially correct. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Electric Conductivity and Density of Sodium-Rubidium and Sodium
Electric Conductivity and Density of Sodium-Rubidium and Sodium-Cesium Acetate Molten Mixtures Dante Leonesi, Augusto Cingolani, and Gianfrancesco Berchiesi Istituto Chimico della Universitä degli Studi, 1-62032 Camerino (Z. Naturforsch. 31a, 1609-1613 [1976]; received April 16, 1976) The electric conductivities, densities and activation energies of RbC2Hs02 and CsC2H302 pure salts and (Na, Rb) C2H302 and (Na, Cs) C2H302 molten mixtures are reported. Deviations from addivity for the Fm, Ae and values are explained. The results for X2H302 where X=Na, K, Rb, Cs and for (Na, X) C2H302, where X = K, Rb, Cs are discussed. Several properties of the alkali metal acetates carrying a Pt-contact for sensing the surface of the were recently investigated by a number of authors1-8. melt in (a), a microamperometer (i) and finally a In particular, the enthalpies and entropies of fusion metallic cover (1) acting as level reference of the and the cryoscopic constants were determined by height before the measurements. The apparatus was our group5_8. More information on the electric calibrated with NaN03 by employing the density data given in 9. conductivities, densities and activation energies at various temperatures for pure RbC2H302 and CsC2H302 and for the mixtures (Na, Rb) C2H302 and (Na, Cs) C2H302 is given in the present paper together with some considerations deduced by com- paring the results of the mixtures (Na, X) C2H302, where X = K, Rb, Cs. Experimental C. Erba RP Sodium Acetate and Nitrate (m. p. 601.0 K and 579.2 K, respectively), K & K 99% Rubidium Acetate (m. p. 514.0 K) and Merck "suprapur" Cesium acetate (m. -

1. Give the Correct Names for Each of the Compounds Listed Below. A
1. Give the correct names for each of the compounds listed below. a) NaCl sodium chloride n) ZrS2 zirconium sulfide b) FrBr francium bromide o) AgI silver iodide c) KF potassium fluoride p) BaSe barium selenide d) RaS radium sulfide q) MgO magnesium oxide e) LiI lithium iodide r) LaBr3 lanthanum bromide f) Li3N lithium nitride s) Sr3N2 strontium nitride g) AlBr3 aluminum bromide t) Cd3As2 cadmium arsenide h) CdCl2 cadmium chloride u) Rb2Se rubidium selenide i) K2O potassium oxide v) Rb3N rubidium nitride j) InF3 indium fluoride w) BaF2 barium fluoride k) ZnO zinc oxide x) ZrTe2 zirconium telluride l) Y2O3 yttrium oxide y) Cs3P cesium phosphide m) CaTe calcium telluride z) Y2O3 yttrium oxide 2. Write the correct chemical formula for each of the following compounds. a) potassium bromide KBr n) potassium nitride K3N b) zinc bromide ZnBr2 o) aluminum bromide AlBr3 c) lithium iodide LiI p) zinc phosphide Zn3P2 d) scandium chloride ScCl3 q) magnesium sulfide MgS e) magnesium chloride MgCl2 r) hafnium chloride HfCl4 f) magnesium oxide MgO s) barium sulfide BaS g) hydrogen sulfide H2S t) tantalum oxide Ta2O5 h) gallium iodide GaI3 u) zirconium nitride Zr3N4 i) sodium oxide Na2O v) potassium selenide K2Se j) magnesium selenide MgSe w) germanium fluoride GeF4 k) calcium fluoride CaF2 x) francium phosphide Fr3P l) aluminum oxide Al2O3 y) zinc arsenide Zn3As2 m) beryllium chloride BeCl2 z) scandium telluride Sc2Te3 L. h. s. – Chemistry – Nomenclature – Answers – Page 1 3. Give the correct names for each of the compounds listed below. a) CaSO4 calcium -

United States Patent Office Patented Sept
3,205,257 United States Patent Office Patented Sept. 7, 1965 2 diols one can mention ethylene glycol, propylene glycol, 3,205,257 tetramethylene diol, decamethylene diol, pentamethylene BES9-(2-CYANOALKYL)FLUOREN-9.YLALKANE COMPOUND diol, hexamethylene diol, and the like. In producing the Henry E. Fritz, South Charleston, and David W. Peck, bis(9-fluorenyl)alkanes the concentration of diol in the Charleston, W. Va., assignors to Union Carbide Corpo charge can vary from about 0.1 mole or less to about 10 ration, a corporation of New York moles or more, per mole of fluorene compound charged, No Drawing. Fied Sept. 10, 1962, Ser. No. 222,694 with from about 0.5 to about 2.0 mole of diol per mole of 5 Caims. (CI. 260-465) fluorene compound being preferred. AS Stated above, an alkali metal hydroxide, such as This invention relates to novel fluorene derivatives. O lithium hydroxide, sodium hydroxide, potassium hydrox More particularly, this invention relates to bis9-(sub ide, and the like, is employed as a catalyst in producing stituted alkyl)fluoren-9-ylalkanes of the formula: the bis(9-fluorenyl)alkanes, with potassium hydroxide be ing preferred. The amount of alkali metal hydroxide used can vary from about 0.1 mole or less to about 1.0 5 mole or more per mole of fluorene compound charged, with from about 0.2 to about 0.5 mole of alkali metal hy -N droxide per mole of fluorene compound being preferred. ZCHRCH CnH2n CHCERZ, The reaction of diol with fluorene is conducted at a tem wherein it is an integer having a valve of from 2 to 10 perature of from about 175 C. -

(12) United States Patent (10) Patent No.: US 9,109,080 B2 Drake Et Al
US009 109080B2 (12) United States Patent (10) Patent No.: US 9,109,080 B2 Drake et al. (45) Date of Patent: *Aug. 18, 2015 (54) CROSS-LINKED ORGANIC POLYMER FOREIGN PATENT DOCUMENTS COMPOSITIONS AND METHODS FOR CONTROLLING CROSS-LINKING EP O251357 B1 1, 1988 REACTION RATE AND OF MODIFYING GB 2185114 7, 1987 SAME TO ENHANCE PROCESSABILITY (Continued) (71) Applicant: Delsper LP, Kulpsville, PA (US) OTHER PUBLICATIONS (72) Inventors: Kerry A. Drake, Red Hill, PA (US); Andrew F. Nordquist, Whitehall, PA Ladacki et al., “Studies of the Variations in Bond Dissociation Ener (US); Sudipto Das, Norristown, PA gies of Aromatic Compounds. I. Mono-bromo-aryles.” Proc. R. Soc. (US); William F. Burgoyne, Jr., Lond. a, r219, pp. 341-253 (1953). Bethlehem, PA (US); Le Song, Chalfont, (Continued) PA (US); Shawn P. Williams, North Wales, PA (US); Rodger K. Boland, Primary Examiner — Robert S. Loewe Levittown, PA (US) (74) Attorney, Agent, or Firm — Flaster/Greenberg P.C. (73) Assignee: Delsper LP, Kulpsville, PA (US) (*) Notice: Subject to any disclaimer, the term of this (57) ABSTRACT patent is extended or adjusted under 35 The invention includes a cross-linking composition compris U.S.C. 154(b) by 0 days. ing a cross-linking compound and a cross-linking reaction additive selected from an organic acid and/or an acetate com This patent is Subject to a terminal dis pound, wherein the cross-linking compound has the structure claimer. according to formula (IV): (21) Appl. No.: 14/059,064 (22) Filed: Oct. 21, 2013 (IV) (65) Prior Publication Data US 2014/0284.850 A1 Sep. -

Download Product List
Promotional Products till stocks last Know more CAS MPN SAP CODE NAME PACK 7440-50-8 039682.22 ALF-039682-22 Copper powder, -100 mesh, 99.999% (metals basis) 100GR 151-21-3 J67606.36 ALF-J67606-36 Sodium n-dodecyl sulfate, 99%, Molecular Biology Grade 500GR 38628-51-2 A15081.22 ALF-A15081-22 3-(4-Carboxyphenyl)propionic acid, 98% 100GR 503-17-3W A13823.22 ALF-A13823-22 2-Butyne, 98% 100GR 7440-06-4 042370.FI ALF-042370-FI Platinum foil, 0.127mm (0.005in) thick, Premion|r, 99.99% (metals basis) 50x50MM 7440-67-7 000418.36 ALF-000418-36 Zirconium powder, -325 mesh, packaged in water 500GR 7440-06-4 012076.06 ALF-012076-06 Platinum black 5GR Tungsten powder, APS 4-6 micron, Puratronic|r, 99.999% (metals basis), 7440-33-7 039691.18 ALF-039691-18 50GR Mo <1ppm 7789-17-5 035729.30 ALF-035729-30 Cesium iodide, ultra dry, 99.998% (metals basis) 250GR 7440-21-3 012681.36 ALF-012681-36 Silicon powder, crystalline,-325 mesh, 99.5% (metals basis) 500GR 203927-98-4 H55817.03 ALF-H55817-03 9,9-Di-n-hexylfluorene-2,7-diboronic acid, 97% 1GR 943-73-7 H27295.14 ALF-H27295-14 L-Homophenylalanine, 98% 25GR 112022-83-0 L14582.AC ALF-L14582-AC (R)-2-Methyl-CBS-oxazaborolidine, 1M soln. in toluene 25ML 775-16-6 A13426.22 ALF-A13426-22 1-Benzyl-3-pyrrolidinone, 98% 100GR 563-67-7 012890.18 ALF-012890-18 Rubidium acetate, 99.8% (metals basis) 50GR 7439-88-5 011430.BU ALF-011430-BU Iridium wire, 0.5mm (0.02in) dia, 99.8% (metals basis) 25CM 105-55-5 A17143.0B ALF-A17143-0B N,N’-Diethylthiourea, 98% 1000GR 13292-46-1 J60836.14 ALF-J60836-14 Rifampin, Molecular -

United States Patent Office Ratiented Sept
2,850,528 United States Patent Office Ratiented Sept. 2, 1958 2 valent elements, particularly the alkali and alkaline earth metals, especially sodium. Thus, a particularly pre 2,850,528 ferred compound of my invention is a-sodio-sodium ace ORGANOMETALLIC COMPOUNDS tate. In certain instances, one or both of the remaining hydrogen atoms on the ox-carbon atom can be substituted Rex D. Closson, Northville, Mich., assignor to Ethyl with lower alkyl radicals, especially those having 3 carbon Corporation, New York, N. Y., a corporation of Del atoms or less, and these compounds will likewise exhibit aware, w the superior characteristics of the structure containing No Drawing. Application June 21, 1954 only 2 carbon atoms. Serial No. 438,357 The novel compositions of my invention have the char acteristic of being stable under atmospheric conditions 5 Claims. (C. 260-54) and at elevated temperatures, especially the o-sodio-so dium acetate. The stability of the O-metallo-metallic acetates surprisingly is superior to that of the a-sodio This invention relates to novel compositions of matter sodium caproate and the ox-sodio-ethyl acetate compounds and in particular to novel organometallic compounds in of the prior art. Another particular advantage of this. which the ce-carbon atom of a metal salt of an organic invention is that it provides o-metalio-metallic organic acid is substituted with a metal. salts essentially free of other organometallic compounds Certain o-metallic substituted organic salts have been which cannot be readily separated therefrom and hinder, set forth in the literature. In particular, c-sodio-sodium. -

Application of Group I Metal Adduction to the Separation of Steroids by Traveling Wave Ion Mobility Spectrometry Alana L
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications -- Chemistry Department Published Research - Department of Chemistry 2018 Application of Group I Metal Adduction to the Separation of Steroids by Traveling Wave Ion Mobility Spectrometry Alana L. Rister University of Nebraska–Lincoln, [email protected] Tiana L. Martin University of Nebraska–Lincoln Eric D. Dodds University of Nebraska-Lincoln, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/chemfacpub Part of the Analytical Chemistry Commons, Medicinal-Pharmaceutical Chemistry Commons, and the Other Chemistry Commons Rister, Alana L.; Martin, Tiana L.; and Dodds, Eric D., "Application of Group I Metal Adduction to the Separation of Steroids by Traveling Wave Ion Mobility Spectrometry" (2018). Faculty Publications -- Chemistry Department. 147. http://digitalcommons.unl.edu/chemfacpub/147 This Article is brought to you for free and open access by the Published Research - Department of Chemistry at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications -- Chemistry Department by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Rister et al. in J. Am. Soc. Mass Spectrom. (2018) 1 Published in Journal of the American Society for Mass Spectrometry (2018) digitalcommons.unl.edu doi 10.1007/s13361-018-2085-9 Copyright © 2018 American Society for Mass Spectrometry. Used by permission. Presented at the 33rd Asilomar Conference, Metabolomics in Translational and Clinical Research. Submitted 1 May 2018; revised 9 August 2018; accepted 10 August 2018; published 9 November 2018. Supplementary material follows the References. Application of Group I Metal Adduction to the Separation of Steroids by Traveling Wave Ion Mobility Spectrometry Alana L. -

Electric Conductivity and Density of Sodium-Rubidium and Sodium-Cesium Acetate Molten Mixtures
Electric Conductivity and Density of Sodium-Rubidium and Sodium-Cesium Acetate Molten Mixtures Dante Leonesi, Augusto Cingolani, and Gianfrancesco Berchiesi Istituto Chimico della Universitä degli Studi, 1-62032 Camerino (Z. Naturforsch. 31a, 1609-1613 [1976]; received April 16, 1976) The electric conductivities, densities and activation energies of RbC2Hs02 and CsC2H302 pure salts and (Na, Rb) C2H302 and (Na, Cs) C2H302 molten mixtures are reported. Deviations from addivity for the Fm, Ae and values are explained. The results for X2H302 where X=Na, K, Rb, Cs and for (Na, X) C2H302, where X = K, Rb, Cs are discussed. Several properties of the alkali metal acetates carrying a Pt-contact for sensing the surface of the were recently investigated by a number of authors1-8. melt in (a), a microamperometer (i) and finally a In particular, the enthalpies and entropies of fusion metallic cover (1) acting as level reference of the and the cryoscopic constants were determined by height before the measurements. The apparatus was our group5_8. More information on the electric calibrated with NaN03 by employing the density data given in 9. conductivities, densities and activation energies at various temperatures for pure RbC2H302 and CsC2H302 and for the mixtures (Na, Rb) C2H302 and (Na, Cs) C2H302 is given in the present paper together with some considerations deduced by com- paring the results of the mixtures (Na, X) C2H302, where X = K, Rb, Cs. Experimental C. Erba RP Sodium Acetate and Nitrate (m. p. 601.0 K and 579.2 K, respectively), K & K 99% Rubidium Acetate (m. p. 514.0 K) and Merck "suprapur" Cesium acetate (m. -

(12) United States Patent (10) Patent No.: US 6,458,915 B1 Quillen (45) Date of Patent: Oct
USOO6458915B1 (12) United States Patent (10) Patent No.: US 6,458,915 B1 Quillen (45) Date of Patent: Oct. 1, 2002 (54) PROCESS FOR PRODUCING POLY (1,4- 5,907,026 A 5/1999 Factor et al. CYCLOHEXYLENEDIMETHYLENE 14- 5,939,519 A 8/1999 Brunelle CYCLOHEXANEDICARBOXYLATE) AND 5,986,040 A 11/1999 Patel et al. THEREFROM FOREIGN PATENT DOCUMENTS (75) Inventor: Donna Rice Quillen, Kingsport, TN EP O 902 052 A1 3/1999 US (US) OTHER PUBLICATIONS (73) ASSignee: ity Company, E.V. Martin and C.J. Kibler, pp. 83-134, in “Man-Made gSpOrl, Fibers: Science and Technology”, Vol. III, edited by Mark, (*) Notice: Subject to any disclaimer, the term of this Atlas and Cernia, 1968. patent is extended or adjusted under 35 Wilfong in J. Polymer Sci., vol. 54,385-410 (1961). U.S.C. 154(b) by 0 days. Primary Examiner Terressa M. Boykin (74) Attorney, Agent, or Firm-Cheryl J. Tubach; Bernard (21) Appl. No.: 09/764,931 J. Graves, Jr. (22) Filed: Jan. 18, 2001 (57) ABSTRACT Related U.S. Application Data In a proceSS for producing a reactor grade polyester, a (60) Provisional application No. 60/240,432, filed on Oct. 13, poly(1, 4-cyclohe Xylene dimethylene 1,4- 2000. cyclohexanedicarboxylate) has a reduced amount of isomer 51) Int. Cl."7 ................................................ C08G 64/00 1Zation off theth trans-ISOmer to thehe cis-ISOmercis-i off 1,4-1.4 (52) U.S. Cl. ........................ 528/272; 528/176; 528/271 dimethylcyclohexanedicarboxylate and an increased (58) Field of Search ................................. 528/176,271, polymerization rate by the addition of a phosphorus 528/272 containing compound to the reaction process.