Photoreceptor-Induced RPE Phagolysosomal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Association Study of Increasing Suicidal Ideation During Antidepressant Treatment in the GENDEP Project
The Pharmacogenomics Journal (2012) 12, 68–77 & 2012 Macmillan Publishers Limited. All rights reserved 1470-269X/12 www.nature.com/tpj ORIGINAL ARTICLE Genome-wide association study of increasing suicidal ideation during antidepressant treatment in the GENDEP project NPerroud1,2,3,RUher1, Suicidal thoughts during antidepressant treatment have been the focus of 1 2,3,4 several candidate gene association studies. The aim of the present genome- MYM Ng , M Guipponi , wide association study was to identify additional genetic variants involved in 5 6 JHauser, N Henigsberg , increasing suicidal ideation during escitalopram and nortriptyline treatment. WMaier7,OMors8, A total of 706 adult participants of European ancestry, treated for major M Gennarelli9, M Rietschel10, depression with escitalopram or nortriptyline over 12 weeks in the Genome- DSouery11, MZ Dernovsek12, Based Therapeutic Drugs for Depression (GENDEP) study were genotyped 13 14 with Illumina Human 610-Quad Beadchips (Illumina, San Diego, CA, USA). AS Stamp ,MLathrop , A total of 244 subjects experienced an increase in suicidal ideation AFarmer1, G Breen1,KJAitchison1, during follow-up. The genetic marker most significantly associated with CM Lewis1,IWCraig1 increasing suicidality (8.28 Â 10À7) was a single-nucleotide polymorphism and P McGuffin1 (SNP; rs11143230) located 30 kb downstream of a gene encoding guanine deaminase (GDA) on chromosome 9q21.13. Two suggestive drug-specific 1MRC Social, Genetic and Developmental Psychiatry associations within KCNIP4 (Kv channel-interacting protein 4; chromosome Centre, Institute of Psychiatry, King’s College 4p15.31) and near ELP3 (elongation protein 3 homolog; chromosome London, London, UK; 2Department of Psychiatry, 8p21.1) were found in subjects treated with escitalopram. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
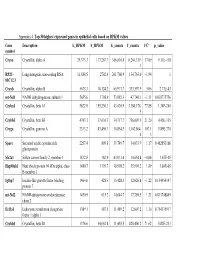
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

A Gene Expression Resource Generated by Genome-Wide Lacz
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 1467-1478 doi:10.1242/dmm.021238 RESOURCE ARTICLE A gene expression resource generated by genome-wide lacZ profiling in the mouse Elizabeth Tuck1,**, Jeanne Estabel1,*,**, Anika Oellrich1, Anna Karin Maguire1, Hibret A. Adissu2, Luke Souter1, Emma Siragher1, Charlotte Lillistone1, Angela L. Green1, Hannah Wardle-Jones1, Damian M. Carragher1,‡, Natasha A. Karp1, Damian Smedley1, Niels C. Adams1,§, Sanger Institute Mouse Genetics Project1,‡‡, James N. Bussell1, David J. Adams1, Ramiro Ramırez-Soliś 1, Karen P. Steel1,¶, Antonella Galli1 and Jacqueline K. White1,§§ ABSTRACT composite of RNA-based expression data sets. Strong agreement was observed, indicating a high degree of specificity in our data. Knowledge of the expression profile of a gene is a critical piece of Furthermore, there were 1207 observations of expression of a information required to build an understanding of the normal and particular gene in an anatomical structure where Bgee had no essential functions of that gene and any role it may play in the data, indicating a large amount of novelty in our data set. development or progression of disease. High-throughput, large- Examples of expression data corroborating and extending scale efforts are on-going internationally to characterise reporter- genotype-phenotype associations and supporting disease gene tagged knockout mouse lines. As part of that effort, we report an candidacy are presented to demonstrate the potential of this open access adult mouse expression resource, in which the powerful resource. expression profile of 424 genes has been assessed in up to 47 different organs, tissues and sub-structures using a lacZ reporter KEY WORDS: Gene expression, lacZ reporter, Mouse, Resource gene. -

The Role of High Density Lipoprotein Compositional and Functional Heterogeneity in Metabolic Disease
The role of high density lipoprotein compositional and functional heterogeneity in metabolic disease By Scott M. Gordon B.S. State University of New York College at Brockport October, 2012 A Dissertation Presented to the Faculty of The University of Cincinnati College of Medicine in partial fulfillment of the requirements for the Degree of Doctor of Philosophy from the Pathobiology and Molecular Medicine graduate program W. Sean Davidson Ph.D. (Chair) David Askew Ph.D. Professor and Thesis Chair Professor Department of Pathology Department of Pathology University of Cincinnati University of Cincinnati Francis McCormack M.D. Gangani Silva Ph.D. Professor Assistant Professor Department of Pathology Department of Pathology University of Cincinnati University of Cincinnati Jason Lu Ph.D. Assistant Professor Division of Bioinformatics Cincinnati Children’s Hospital i Abstract High density lipoproteins (HDL) are complexes of phospholipid, cholesterol and protein that circulate in the blood. Epidemiological studies have demonstrated a strong inverse correlation between plasma levels of HDL associated cholesterol (HDL-C) and the incidence of cardiovascular disease (CVD). Clinically, HDL-C is often measured and used in combination with low density lipoprotein cholesterol (LDL-C) to assess overall cardiovascular health. HDL have been shown to possess a wide variety of functional attributes which likely contribute to this protection including anti-inflammatory and anti- oxidative properties and the ability to remove excess cholesterol from peripheral tissues and deliver it to the liver for excretion, a process known as reverse cholesterol transport. This functional diversity might be explained by the complexity of HDL composition. Recent studies have taken advantage of advances in mass spectrometry technologies to characterize the proteome of total HDL finding that over 50 different proteins can associate with these particles. -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Genomic Aberrations Associated with Erlotinib Resistance in Non-Small Cell Lung Cancer Cells
ANTICANCER RESEARCH 33: 5223-5234 (2013) Genomic Aberrations Associated with Erlotinib Resistance in Non-small Cell Lung Cancer Cells MASAKUNI SERIZAWA1, TOSHIAKI TAKAHASHI2, NOBUYUKI YAMAMOTO2,3 and YASUHIRO KOH1 1Drug Discovery and Development Division, Shizuoka Cancer Center Research Institute, Sunto-gun, Shizuoka, Japan; 2Division of Thoracic Oncology, Shizuoka Cancer Center Hospital, Sunto-gun, Shizuoka, Japan; 3Third Department of Internal Medicine, Wakayama Medical University, Kimiidera, Wakayama, Japan Abstract. Background/Aim: Mechanisms of resistance to mutations develop resistance, usually within one year of epidermal growth factor receptor (EGFR)-tyrosine kinase treatment. Therefore, there is an urgent need to elucidate the inhibitors (TKIs) in non-small cell lung cancer (NSCLC) underlying mechanisms of resistance in such tumors to are not fully-understood. In this study we aimed to overcome this obstacle (11-14, 17, 24). Recent studies elucidate remaining unknown mechanisms using erlotinib- suggest that mechanisms of acquired resistance to EGFR- resistant NSCLC cells. Materials and Methods: We TKIs can be categorized into three groups: occurrence of performed array comparative genomic hybridization genetic alterations, activation of downstream pathways via (aCGH) to identify genomic aberrations associated with bypass signaling, and phenotypic transformation (15, 16, 21); EGFR-TKI resistance in erlotinib-resistant PC-9ER cells. therapeutic strategies to overcome these resistance Real-time polymerase chain reaction (PCR) and mechanisms are under development. However, although the immunoblot analyses were performed to confirm the results causes of acquired resistance to EGFR-TKIs have been of aCGH. Results: Among the five regions with copy investigated, in more than 30% of patients with acquired number gain detected in PC-9ER cells, we focused on resistance to EGFR-TKI treatment, the mechanisms remain 22q11.2-q12.1 including v-crk avian sarcoma virus CT10 unknown (15). -

Research Article Characterization of the Equine Skeletal Muscle
McGivney et al. BMC Genomics 2010, 11:398 http://www.biomedcentral.com/1471-2164/11/398 RESEARCH ARTICLE Open Access CharacterizationResearch article of the equine skeletal muscle transcriptome identifies novel functional responses to exercise training Beatrice A McGivney1, Paul A McGettigan1, John A Browne1, Alexander CO Evans1,3, Rita G Fonseca2, Brendan J Loftus3, Amanda Lohan3, David E MacHugh1,3, Barbara A Murphy1, Lisa M Katz2 and Emmeline W Hill*1 Abstract Background: Digital gene expression profiling was used to characterize the assembly of genes expressed in equine skeletal muscle and to identify the subset of genes that were differentially expressed following a ten-month period of exercise training. The study cohort comprised seven Thoroughbred racehorses from a single training yard. Skeletal muscle biopsies were collected at rest from the gluteus medius at two time points: T1 - untrained, (9 ± 0.5 months old) and T2 - trained (20 ± 0.7 months old). Results: The most abundant mRNA transcripts in the muscle transcriptome were those involved in muscle contraction, aerobic respiration and mitochondrial function. A previously unreported over-representation of genes related to RNA processing, the stress response and proteolysis was observed. Following training 92 tags were differentially expressed of which 74 were annotated. Sixteen genes showed increased expression, including the mitochondrial genes ACADVL, MRPS21 and SLC25A29 encoded by the nuclear genome. Among the 58 genes with decreased expression, MSTN, a negative regulator of muscle growth, had the greatest decrease. Functional analysis of all expressed genes using FatiScan revealed an asymmetric distribution of 482 Gene Ontology (GO) groups and 18 KEGG pathways. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

AP1B1 Rabbit Polyclonal Antibody – TA323221 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA323221 AP1B1 Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: IHC Recommended Dilution: ELISA: 1:1000-5000, IHC: 1:25-100 Reactivity: Human, Mouse, Rat Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: Synthetic peptide corresponding to a region derived from 18-30 amino acids of Human Adapter-related protein complex 1 subunit beta-1 Formulation: PBS pH7.3, 0.05% NaN3, 50% glycerol Concentration: lot specific Purification: Antigen affinity purification Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: adaptor related protein complex 1 beta 1 subunit Database Link: NP_001118 Entrez Gene 11764 MouseEntrez Gene 29663 RatEntrez Gene 162 Human Q10567 Background: Adaptor protein complex 1 is found at the cytoplasmic face of coated vesicles located at the Golgi complex; where it mediates both the recruitment of clathrin to the membrane and the recognition of sorting signals within the cytosolic tails of transmembrane receptors. This complex is a heterotetramer composed of two large; one medium; and one small adaptin subunit. The protein encoded by this gene serves as one of the large subunits of this complex and is a member of the adaptin protein family. This gene is a candidate meningioma gene. Alternative splicing results in multiple transcript variants. Synonyms: ADTB1; AP105A; BAM22; CLAPB2 Protein Pathways: Lysosome This product is to be used for laboratory only. -

Method for Diagnosing Non-Small Cell Lung Cancers
(19) TZZ Z _ T (11) EP 2 270 221 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 05.01.2011 Bulletin 2011/01 C12Q 1/68 (2006.01) C12N 15/12 (2006.01) G01N 33/50 (2006.01) C12N 15/11 (2006.01) (2006.01) (2006.01) (21) Application number: 10010329.0 A61P 35/00 A61K 39/00 (22) Date of filing: 22.09.2003 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Nakamura, Yusuke HU IE IT LI LU MC NL PT RO SE SI SK TR Yokohama-shi Kanagawa 225-0011 (JP) (30) Priority: 30.09.2002 US 414673 P • Daigo, Yataro 28.02.2003 US 451374 P Yokohama-shi 28.04.2003 US 466100 P Kanagawa 222-0031 (JP) • Nakatsuru, Shuichi (62) Document number(s) of the earlier application(s) in Saitama-shi accordance with Art. 76 EPC: Saitama 338-0002 (JP) 06022167.8 / 1 743 947 03753941.8 / 1 551 998 (74) Representative: Vossius & Partner Siebertstrasse 4 (71) Applicant: Oncotherapy Science, Inc. 81675 München (DE) Kawasaki-shi Kanagawa 213-0012 (JP) Remarks: This application was filed on 23-09-2010 as a divisional application to the application mentioned under INID code 62. (54) Method for diagnosing non-small cell lung cancers (57) Disclosed are methods for detecting non-small cancerous tissues are provided. Also disclosed are meth- cell lung cancer using differentially expressed genes. ods of identifying compounds for treating and preventing Furthermore, novel human genes whose expression is non-small cell lung cancer. -

Congenital Cataracts Due to a Novel 2‑Bp Deletion in CRYBA1/A3
1614 MOLECULAR MEDICINE REPORTS 10: 1614-1618, 2014 Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3 JING ZHANG1, YANHUA ZHANG1, FANG FANG1, WEIHONG MU1, NING ZHANG2, TONGSHUN XU3 and QINYING CAO1 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital; 2Department of Cardiology, The Second Hospital of Hebei Medical University; 3Department of Surgery, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, P.R. China Received September 22, 2013; Accepted April 11, 2014 DOI: 10.3892/mmr.2014.2324 Abstract. Congenital cataracts, which are a clinically and located in the eye lens. The major human crystallins comprise genetically heterogeneous group of eye disorders, lead to 90% of protein in the mature lens and contain two different visual impairment and are a significant cause of blindness superfamilies: the small heat‑shock proteins (α-crystallins) in childhood. A major proportion of the causative mutations and the βγ-crystallins. for congenital cataracts are found in crystallin genes. In the In this study a functional candidate approach was used present study, a novel deletion mutation (c.590-591delAG) in to investigate the known crystallin genes, including CRYAA, exon 6 of CRYBA1/A3 was identified in a large family with CRYAB, CRYBA1/A3, CRYBB1, CRYBB2, CRYGC, CRYGD autosomal dominant congenital cataracts. An increase in and CRYGS, in which a major proportion of the mutations local hydrophobicity was predicted around the mutation site; identified in a large family with congenital cataracts were however, further studies are required to determine the exact found. effect of the mutation on βA1/A3-crystallin structure and function. To the best of our knowledge, this is the first report Subjects and methods of an association between a frameshift mutation in exon 6 of CRYBA1/A3 and congenital cataracts.