Investigation on Genetic Modifiers of Age at Onset of Major Depressive Disorder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Meta-Analysis and Inding from Seoul Breast Cancer Study (SEBCS)
The Pharmacogenomics Journal https://doi.org/10.1038/s41397-018-0016-6 ARTICLE Associations between genetic polymorphisms of membrane transporter genes and prognosis after chemotherapy: meta-analysis and finding from Seoul Breast Cancer Study (SEBCS) 1 1 1 1 2 3 4 Ji-Eun Kim ● Jaesung Choi ● JooYong Park ● Chulbum Park ● Se Mi Lee ● Seong Eun Park ● Nan Song ● 5 6 4,7 8 1,4,9 10 Seokang Chung ● Hyuna Sung ● Wonshik Han ● Jong Won Lee ● Sue K. Park ● Mi Kyung Kim ● 4,7 9,11 1,4,9,12 1,4,9 Dong-Young Noh ● Keun-Young Yoo ● Daehee Kang ● Ji-Yeob Choi Received: 7 June 2017 / Revised: 13 October 2017 / Accepted: 4 December 2017 © Macmillan Publishers Limited, part of Springer Nature 2018 Abstract Membrane transporters can be major determinants of the pharmacokinetic profiles of anticancer drugs. The associations between genetic variations of ATP-binding cassette (ABC) and solute carrier (SLC) genes and cancer survival were investigated through a meta-analysis and an association study in the Seoul Breast Cancer Study (SEBCS). Including the SEBCS, the meta-analysis was conducted among 38 studies of genetic variations of transporters on various cancer survivors. 1234567890();,: The population of SEBCS consisted of 1 338 breast cancer patients who had been treated with adjuvant chemotherapy. A total of 7 750 SNPs were selected from 453 ABC and/or SLC genes typed by an Affymetrix 6.0 chip. ABCB1 rs1045642 was associated with poor progression-free survival in a meta-analysis (HR = 1.33, 95% CI: 1.07–1.64). ABCB1, SLC8A1, and SLC12A8 were associated with breast cancer survival in SEBCS (Pgene < 0.05). -

Serotonin Receptor 2A (HTR2A) Gene Polymorphism Predicts Treatment Response to Venlafaxine XR in Generalized Anxiety Disorder
The Pharmacogenomics Journal (2013) 13, 21–26 & 2013 Macmillan Publishers Limited. All rights reserved 1470-269X/13 www.nature.com/tpj ORIGINAL ARTICLE Serotonin receptor 2A (HTR2A) gene polymorphism predicts treatment response to venlafaxine XR in generalized anxiety disorder FW Lohoff1,2, TD Aquino1, Generalized anxiety disorder (GAD) is a chronic psychiatric disorder with 2 2 significant morbidity and mortality. Antidepressant drugs are the preferred S Narasimhan , PK Multani , choice for treatment; however, treatment response is often variable. Several 1 1 B Etemad and K Rickels studies in major depression have implicated a role of the serotonin receptor gene (HTR2A) in treatment response to antidepressants. We tested the 1Mood & Anxiety Disorders Section, Department of Psychiatry, University of Pennsylvania School of hypothesis that the genetic polymorphism rs7997012 in the HTR2A gene Medicine, Philadelphia, PA, USA and predicts treatment outcome in GAD patients treated with venlafaxine XR. 2Department of Psychiatry, Psychiatric Treatment response was assessed in 156 patients that participated in a Pharmacogenetics Laboratory, Center for 6-month open-label clinical trial of venlafaxine XR for GAD. Primary analysis Neurobiology and Behavior, University of Pennsylvania School of Medicine, Philadelphia, included Hamilton Anxiety Scale (HAM-A) reduction at 6 months. Secondary PA, USA outcome measure was the Clinical Global Impression of Improvement (CGI-I) score at 6 months. Genotype and allele frequencies were compared between Correspondence: groups using w2 contingency analysis. The frequency of the G-allele differed Dr FW Lohoff, Department of Psychiatry, significantly between responders (70%) and nonresponders (56%) at Translational Research Laboratory, Center for Neurobiology and Behavior, University of 6 months (P ¼ 0.05) using the HAM-A scale as outcome measure. -

New Products JULY 2013
R&D Systems Tools for Cell Biology Research™ New Products JULY 2013 GMP-grade Recombinant Proteins Contents R&D Systems now offers GMP-grade cytokines and growth factors for research and further manu- facturing applications where current Good Manufacturing Practices (GMP) are required. GMP-grade Recombinant Proteins 2 proteins are manufactured in our ISO-certified facility in compliance with relevant guidelines1 and are produced with extensive documentation at every stage of development from cell culture to final fill and Quantikine® ELISA Kits 3 formulation. GMP-grade Recombinant Human IL-6 and Recombinant Human TNF-a have recently been added to our line of GMP-grade proteins. Additional GMP-grade proteins will be available in the next several months. For an up-to-date product listing or additional information, please visit our website at Luminex® Screening Assays 4 www.RnDSystems.com/GMP. Luminex® Performance Assays 4-5 Features New GMP Proteins ✓ Extensive documentation at every stage of development ProtEin SOURCE Catalog # SIZE Polyclonal Antibodies 6-7 ✓ Documentation of lot-to-lot consistency and traceability Human IL-6 E. coli 206-GMP-010 10 µg Monoclonal Antibodies 7-8 of materials used 206-GMP-050 50 µg ✓ Rigorous quality control using stringent analytical 206-GMP-01M 1 mg processes Biotinylated Antibodies 8 Human TNF-a/ E. coli 210-GMP-010 10 µg ✓ Proven formulations to ensure consistent reconstitution TNFSF1A and results 210-GMP-050 50 µg Antibody Controls 8 210-GMP-01M 1 mg ELISpot Kits & Development Modules 8 30000 60 Fluorokine® Flow Cytometry Kits 8 25000 17348 ) 2 50 20000 Fluorochrome-labeled Antibodies 9 40 8673 DuoSet® ELISA and DuoSet IC ELISA 15000 Development Systems 10 30 Peak Intensity Peak 10000 20 Cell-Based ELISA Assay Kits 10 5000 17565 (Mean RFU x10 Viability Cell 8780 10 Parameter Assay Kits 10 0 0 6000 9000 12000 15000 18000 21000 24000 -3 -2 -1 0 1 10 10 10 10 10 Apoptosis Detection 10 Mass Charge Ratio Recombinant Human TNF-α GMP (ng/mL) MALDI-TOF Analysis of GMP-grade Recombinant Human TNF-a. -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Supplemental Figure 1. Vimentin
Double mutant specific genes Transcript gene_assignment Gene Symbol RefSeq FDR Fold- FDR Fold- FDR Fold- ID (single vs. Change (double Change (double Change wt) (single vs. wt) (double vs. single) (double vs. wt) vs. wt) vs. single) 10485013 BC085239 // 1110051M20Rik // RIKEN cDNA 1110051M20 gene // 2 E1 // 228356 /// NM 1110051M20Ri BC085239 0.164013 -1.38517 0.0345128 -2.24228 0.154535 -1.61877 k 10358717 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 /// BC 1700025G04Rik NM_197990 0.142593 -1.37878 0.0212926 -3.13385 0.093068 -2.27291 10358713 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 1700025G04Rik NM_197990 0.0655213 -1.71563 0.0222468 -2.32498 0.166843 -1.35517 10481312 NM_027283 // 1700026L06Rik // RIKEN cDNA 1700026L06 gene // 2 A3 // 69987 /// EN 1700026L06Rik NM_027283 0.0503754 -1.46385 0.0140999 -2.19537 0.0825609 -1.49972 10351465 BC150846 // 1700084C01Rik // RIKEN cDNA 1700084C01 gene // 1 H3 // 78465 /// NM_ 1700084C01Rik BC150846 0.107391 -1.5916 0.0385418 -2.05801 0.295457 -1.29305 10569654 AK007416 // 1810010D01Rik // RIKEN cDNA 1810010D01 gene // 7 F5 // 381935 /// XR 1810010D01Rik AK007416 0.145576 1.69432 0.0476957 2.51662 0.288571 1.48533 10508883 NM_001083916 // 1810019J16Rik // RIKEN cDNA 1810019J16 gene // 4 D2.3 // 69073 / 1810019J16Rik NM_001083916 0.0533206 1.57139 0.0145433 2.56417 0.0836674 1.63179 10585282 ENSMUST00000050829 // 2010007H06Rik // RIKEN cDNA 2010007H06 gene // --- // 6984 2010007H06Rik ENSMUST00000050829 0.129914 -1.71998 0.0434862 -2.51672 -

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Pharmacogenomic Characterization in Bipolar Spectrum Disorders
pharmaceutics Review Pharmacogenomic Characterization in Bipolar Spectrum Disorders Stefano Fortinguerra 1,2 , Vincenzo Sorrenti 1,2,3 , Pietro Giusti 2, Morena Zusso 2 and Alessandro Buriani 1,2,* 1 Maria Paola Belloni Center for Personalized Medicine, Data Medica Group (Synlab Limited), 35131 Padova, Italy; [email protected] (S.F.); [email protected] (V.S.) 2 Department of Pharmaceutical & Pharmacological Sciences, University of Padova, 35131 Padova, Italy; [email protected] (P.G.); [email protected] (M.Z.) 3 Bendessere™ Study Center, Solgar Italia Multinutrient S.p.A., 35131 Padova, Italy * Correspondence: [email protected] Received: 25 November 2019; Accepted: 19 December 2019; Published: 21 December 2019 Abstract: The holistic approach of personalized medicine, merging clinical and molecular characteristics to tailor the diagnostic and therapeutic path to each individual, is steadily spreading in clinical practice. Psychiatric disorders represent one of the most difficult diagnostic challenges, given their frequent mixed nature and intrinsic variability, as in bipolar disorders and depression. Patients misdiagnosed as depressed are often initially prescribed serotonergic antidepressants, a treatment that can exacerbate a previously unrecognized bipolar condition. Thanks to the use of the patient’s genomic profile, it is possible to recognize such risk and at the same time characterize specific genetic assets specifically associated with bipolar spectrum disorder, as well as with the individual response to the various therapeutic options. This provides the basis for molecular diagnosis and the definition of pharmacogenomic profiles, thus guiding therapeutic choices and allowing a safer and more effective use of psychotropic drugs. Here, we report the pharmacogenomics state of the art in bipolar disorders and suggest an algorithm for therapeutic regimen choice. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -
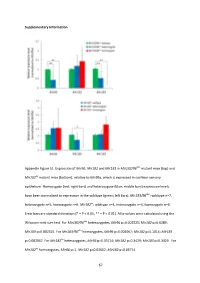
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old.