Draft Genome of the Globally Widespread and Invasive Argentine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Winged Ants (Hymenoptera: Formicidae) Presence in Twigs on the Leaf Litter of Atlantic Forest
Biota Neotropica 19(3): e20180694, 2019 www.scielo.br/bn ISSN 1676-0611 (online edition) Inventory Winged ants (Hymenoptera: Formicidae) presence in twigs on the leaf litter of Atlantic Forest Tae Tanaami Fernandes1 , Rogério R. Silva2, Débora Rodrigues de Souza-Campana2, Otávio Guilherme Morais da Silva2 & Maria Santina de Castro Morini1* 1Universidade de Mogi das Cruzes, Laboratório de Mirmecologia do Alto Tietê, Rua Dr. Cândido Xavier de Almeida e Souza, 200, CEP 08780-911, Mogi das Cruzes, SP, Brasil 2Museu Paraense Emílio Goeldi, Coordenação de Ciências da Terra e Ecologia, Avenida Perimetral, 1901, Terra Firme, CEP 66077-830, Belém, PA, Brasil *Corresponding author: Maria Santina de Castro Morini, e-mail: [email protected] FERNANDES, T. T., SILVA, R. R., SOUZA-CAMPANA, D. R., SILVA, O. G. M., MORINI, M. S. C. Winged ants (Hymenoptera: Formicidae) presence in twigs on the leaf litter of Atlantic Forest. Biota Neotropica 19(3): e20180694. http://dx.doi.org/10.1590/1676-0611-BN-2018-0694 Abstract: In the leaf litter, ants have various nesting resources available, such as live or dead trunks, twigs, leaves, fruits and seeds. On the twigs, there are adults and immature individuals, but also the queen and winged. The production of wings requires time and energy from the colony. The objective of this study was to investigate the presence of winged in ant colonies in twigs on the leaf litter. Our prediction is that the richness and abundance of winged in twigs are the greatest in rainy months. We collected all twigs with ants in 552 plots with 16 m2, totaling 8,832 m2 of leaf litter, in areas located in the Brazilian Atlantic Domain. -

ORIGINAL ARTICLE a Novel Intracellular Mutualistic Bacterium in the Invasive Ant Cardiocondyla Obscurior
The ISME Journal (2016) 10, 376–388 © 2016 International Society for Microbial Ecology All rights reserved 1751-7362/16 www.nature.com/ismej ORIGINAL ARTICLE A novel intracellular mutualistic bacterium in the invasive ant Cardiocondyla obscurior Antonia Klein1,8, Lukas Schrader1,8, Rosario Gil2, Alejandro Manzano-Marín2, Laura Flórez3, David Wheeler5, John H Werren6, Amparo Latorre2,7, Jürgen Heinze1, Martin Kaltenpoth3,4, Andrés Moya2 and Jan Oettler1 1Institut für Zoologie, Universität Regensburg, Regensburg, Germany; 2Institut Canvanilles de Biodiversitat i Biologia Evolutiva (ICBiBE), Parc Cientific de la Universitat de Valencia, Paterna (Valencia), Spain; 3Max Planck Institute for Chemical Ecology, Jena, Germany; 4Johannes Gutenberg University Mainz, Institute for Zoology, Department for Evolutionary Ecology, Mainz, Germany; 5Institute of Fundamental Sciences, Massey University, Palmerston North, New Zealand; 6Department of Biology, University Rochester, Rochester, NY, USA and 7Área de Genómica y Salud de la Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana (FISABIO)–Salud Pública, Valencia, Spain The evolution of eukaryotic organisms is often strongly influenced by microbial symbionts that confer novel traits to their hosts. Here we describe the intracellular Enterobacteriaceae symbiont of theinvasiveantCardiocondyla obscurior, ‘Candidatus Westeberhardia cardiocondylae’. Upon metamorphosis, Westeberhardia is found in gut-associated bacteriomes that deteriorate following eclosion. Only queens maintain Westeberhardia in the ovarian nurse cells from where the symbionts are transmitted to late-stage oocytes during nurse cell depletion. Functional analyses of the streamlined genome of Westeberhardia (533 kb, 23.41% GC content) indicate that neither vitamins nor essential amino acids are provided for the host. However, the genome encodes for an almost complete shikimate pathway leading to 4-hydroxyphenylpyruvate, which could be converted into tyrosine by the host. -

Taxonomy and Distribution of the Argentine Ant, Linepithema Humile (Hymenoptera: Formicidae)
SYSTEMATICS Taxonomy and Distribution of the Argentine Ant, Linepithema humile (Hymenoptera: Formicidae) ALEXANDER L. WILD Department of Entomology, University of California at Davis, Davis, CA 95616 Ann. Entomol. Soc. Am. 97(6): 1204Ð1215 (2004) ABSTRACT The taxonomy of an invasive pest species, the Argentine ant, is reviewed. Linepithema humile (Mayr) 1868 is conÞrmed as the valid name for the Argentine ant. Morphological variation and species boundaries of L. humile are examined, with emphasis on populations from the antÕs native range in southern South America. Diagnoses and illustrations are provided for male, queen, and worker castes. Collection records of L. humile in South America support the idea of a native distribution closely associated with major waterways in lowland areas of the Parana´ River drainage, with recent intro- ductions into parts of Argentina, Brazil, Chile, Colombia, Ecuador, and Peru. KEY WORDS Argentine ant, Linepithema humile, taxonomy, invasive species THE ARGENTINE ANT, Linepithema humile (Mayr) 1868, MCSN, MCZC, MHNG, MZSP, NHMB, NHMW, and is among the worldÕs most successful invasive species. USNM; see below for explanation of abbreviations). This native South American insect has become a cos- Taxonomic confusion over L. humile extends be- mopolitan pest, particularly in the Mediterranean cli- yond museum collections. At least one important mates of North America, Chile, South Africa, Austra- study, seeking to explain Argentine ant population lia, and southern Europe (Suarez et al. 2001). regulation in the native range through phorid para- Argentine ants have been implicated in the decline of sitism (Orr and Seike 1998), initially targeted the native arthropod (Cole et al. 1992) and vertebrate wrong Linepithema species (Orr et al. -

Population Dynamics of Invasive Argentine Ant
BioInvasions Records (2021) Volume 10, Issue 2: 467–476 CORRECTED PROOF Research Article Population dynamics of invasive Argentine ant Linepithema humile Mayr, 1868 (Hymenoptera: Formicidae) haplotypes in Kobe Port, Japan, and implications for the prediction of future dispersal and effective management Yugo Seko1,#,*, Yu Maebara2,#, Naoyuki Nakahama3,4, Takuya Nakamori1, Naoto Ishiwaka5, Yuki Morikawa5, Daisuke Hayasaka1,* and Takuo Sawahata1 1Graduate school of Agriculture, Kindai University, 3327-204, Nakamachi, Nara City, Nara Prefecture, Japan 2Nagoya Branch Office, Nippon Koei Co. Ltd., 1-20-22, Aoi, Naka-ku, Nagoya City, Aichi Prefecture, Japan 3Institute of Natural and Environmental Sciences, University of Hyogo, 6 Yayoigaoka, Sanda City, Hyogo Prefecture, Japan 4Division of Ecological Restoration, The Museum of Nature and Human Activities, 6 Yayoigaoka, Sanda City, Hyogo Prefecture, Japan 5Faculty of Agriculture, Kindai University, 3327-204, Nakamachi, Nara City, Nara Prefecture, Japan Author e-mails: [email protected] (YS), [email protected] (YMa), [email protected] (NN), [email protected] (TN), [email protected] (NI), [email protected] (YMo), [email protected]; [email protected] (DH), [email protected] (TS) #These authors contributed equally *Corresponding authors Citation: Seko Y, Maebara Y, Nakahama N, Nakamori T, Ishiwaka N, Morikawa Y, Abstract Hayasaka D, Sawahata T (2021) Population dynamics of invasive Argentine The Argentine ant Linepithema humile Mayr, native to South America, has been ant Linepithema humile Mayr, 1868 unintentionally introduced worldwide. Although L. humile usually forms an extremely (Hymenoptera: Formicidae) haplotypes in large supercolony all sharing the same haplotype among members in its introduced Kobe Port, Japan, and implications for the ranges, four haplotypes (LH1, LH2, LH3, LH4) with different genetic structures prediction of future dispersal and effective management. -

Genetic Mechanisms Underlying the Evolutionary Success of Eusocial Insects
insects Review (Epi)Genetic Mechanisms Underlying the Evolutionary Success of Eusocial Insects Kayli R. Sieber 1 , Taylor Dorman 1, Nicholas Newell 1 and Hua Yan 1,2,* 1 Department of Biology, University of Florida, Gainesville, FL 32611, USA; kayli.sieber@ufl.edu (K.R.S.); taylor.dorman@ufl.edu (T.D.); nicholas.newell@ufl.edu (N.N.) 2 Center for Smell and Taste, University of Florida, Gainesville, FL 32611, USA * Correspondence: hua.yan@ufl.edu; Tel.: +1-352-273-4983 Simple Summary: Social insects, namely ants, bees, and termites, are among the most numerous and successful animals on Earth. This is due to a variety of features: highly cooperative behavior performed by colony members and their specialization on a variety of tasks. Diverse physiological and behavioral specializations are regulated not only by the genetic system, but also by the epige- netic system which alters gene expressions without modifying the genetic code. This review will summarize recent advancements in such studies in eusocial insects. Abstract: Eusocial insects, such as bees, ants, and wasps of the Hymenoptera and termites of the Blattodea, are able to generate remarkable diversity in morphology and behavior despite being genetically uniform within a colony. Most eusocial insect species display caste structures in which reproductive ability is possessed by a single or a few queens while all other colony members act Citation: Sieber, K.R.; Dorman, T.; as workers. However, in some species, caste structure is somewhat plastic, and individuals may Newell, N.; Yan, H. (Epi)Genetic switch from one caste or behavioral phenotype to another in response to certain environmental cues. -
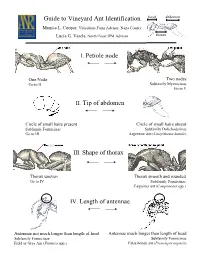
I. Petiole Node II. Tip of Abdomen IV. Length of Antennae Guide to Vineyard Ant Identification III. Shape of Thorax
Guide to Vineyard Ant Identification head abdomen Monica L. Cooper, Viticulture Farm Advisor, Napa County Lucia G. Varela, North Coast IPM Advisor thorax I. Petiole node One Node Two nodes Go to II Subfamily Myrmicinae Go to V II. Tip of abdomen Circle of small hairs present Circle of small hairs absent Subfamily Formicinae Subfamily Dolichoderinae Go to III Argentine Ant (Linepithema humile) III. Shape of thorax Thorax uneven Thorax smooth and rounded Go to IV Subfamily Formicinae Carpenter ant (Camponotus spp.) IV. Length of antennae Antennae not much longer than length of head Antennae much longer than length of head Subfamily Formicinae Subfamily Formicinae Field or Gray Ant (Formica spp.) False honey ant (Prenolepis imparis) head abdomen Petiole with two nodes Subfamily Myrmicinae (V-VIII) thorax V. Dorsal side of Thorax & Antennae One pair of spines on thorax No spines on thorax 12 segmented antennae 10 segmented antennae Go to VI Solenopsis molesta and Solenopsis xyloni VI. Underside of head No brush of bristles Brush of long bristles Go to VII Harvester ants (Pogonomyrmex californicus and P. brevispinosis) VII. Head and Thorax With hairs Without hairs Go to VIII Cardiocondyla mauritanica VIII. Head and Thorax With many parallel furrows Without parallel furrows Profile of thorax rounded Profile of thorax not evenly rounded Pavement ant (Tetramorium “species E”) Pheidole californica Argentine Ant (Linepithema humile), subfamily Dolichoderinae Exotic species 3-4 mm in length Deep brown to light black Move rapidly in distinct trails Feed on honeydew Shallow nests (2 inches from soil surface) Alex Wild Does not bite or sting Carpenter Ant (Camponotus spp.), subfamily Formicinae Large ant: >6 mm in length Dark color with smooth, rounded thorax Workers most active at dusk and night One of most abundant and widespread genera worldwide Generalist scavengers and predators: feed on dead and living insects, nectar, fruit juices and Jack K. -

Hymenoptera: Formicidae: Dolichoderinae) from Colombia
Revista Colombiana de Entomología 37 (1): 159-161 (2011) 159 Scientific note The first record of the genus Gracilidris (Hymenoptera: Formicidae: Dolichoderinae) from Colombia Primer registro del género Gracilidris (Hymenoptera: Formicidae: Dolichoderinae) para Colombia ROBERTO J. GUERRERO1 and CATALINA SANABRIA2 Abstract: The dolichoderine ant genus Gracilidris and its sole species, G. pombero, are recorded for the first time for Colombia from populations from the foothills of the Colombian Amazon basin. Comments and hypotheses about the biogeography of the genus are discussed. Key words: Ants. Biodiversity. Caquetá. Colombian Amazon. Grazing systems. Resumen: El género dolicoderino de hormigas Gracilidris y su única especie, G. pombero, son registrados por primera vez para Colombia, de poblaciones provenientes del piedemonte de la cuenca Amazónica colombiana. Algunos comen- tarios e hipótesis sobre la biogeografía del género son discutidos. Palabras clave: Hormigas. Biodiversidad. Caquetá. Amazonas colombiano. Pasturas ganaderas. Introduction distribution of the genus Gracilidris in South America. We also discuss each of the dolichoderine genera that occur in Currently, dolichoderine ants (Hymenoptera: Formicidae: Colombia. Dolichoderinae) include 28 extant genera (Bolton et al. 2006; Fisher 2009) distributed in four tribes according to Methods the latest higher level classification of the ant subfamily Dolichoderinae (Ward et al. 2010). Eleven of those extant We separated G. pombero specimens from all ants collected genera occur in the New World: Bothriomyrmex, Dolicho- in the project “Amaz_BD: Biodiversidad de los paisajes derus, Liometopum, Tapinoma, and Technomyrmex have a Amazónicos, determinantes socio-económicos y produc- global distribution, while Anillidris, Azteca, Dorymyrmex, ción de bienes y servicios”. This research was carried out in Forelius, Gracilidris, and Linepithema are endemic to the Caquetá department located in the western foothills of the New World. -

Use of an Argentine Ant, Linepithema Humile, Semiochemical to Deliver an Acute Toxicant
insects Article Use of an Argentine Ant, Linepithema humile, Semiochemical to Deliver an Acute Toxicant Benjamin M. Gochnour 1, Daniel R. Suiter 2,*, Jerry W. Davis 3 and Qingguo Huang 4 1 Department of Entomology, University of Georgia, 120 Cedar Street, Athens, GA 30602, USA; [email protected] 2 Department of Entomology, University of Georgia Griffin Camus, 1109 Experiment Street, Griffin, GA 30223, USA 3 Experimental Statistics, College of Agricultural and Environmental Sciences, University of Georgia, 1109 Experiment Street, Griffin, GA 30223, USA; [email protected] 4 Department of Crop and Soil Sciences, University of Georgia Griffin Campus, 1109 Experiment Street, Griffin, GA 30223, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-770-233-6114 Received: 14 October 2018; Accepted: 16 November 2018; Published: 23 November 2018 Abstract: The Argentine ant, Linepithema humile, is an invasive nuisance, agricultural, and ecological pest from South America. In the United States, its primary distribution is in California and the Southeast. The structural pest control industry responds to property owner complaints when this ant’s populations become problematic and a persistent nuisance. Actions taken to control Argentine ants in the urban and suburban environment are typically complaint-driven, and often involve the application of insecticide sprays applied to the outdoor environment by professional pest managers. In California, and elsewhere, spray treatments of various residual insecticides by property owners and pest management professionals has resulted in significant runoff and in subsequent surface water contamination. As a result, an immediate need exists to develop alternative methods of ant control targeted at reducing environmental contamination. -

Diversity of Ants (Hymenoptera: Formicidae) Inside and Outside Hives of the Western Honey Bee Apis Mellifera L
44 ChileanChilean J. Agric. J. Agric. Anim. Anim. Sci., exSci., Agro-Ciencia ex Agro-Ciencia (2020) (2020) 36(1): 36(1):44-51.44-51. https://doi.org/10.29393/CHJAAS36-1D40001 ISSN 0719-3882 print ISSN 0719-3890 online DIVERSITY OF ANTS (HYMENOPTERA: FORMICIDAE) INSIDE AND OUTSIDE HIVES OF THE WESTERN HONEY BEE APIS MELLIFERA L. (HYMENOPTERA: APIDAE), JUJUY, ARGENTINA Gisela Beatriz Ruiz1, 2* and Marcelo Benítez Ahrendts 1, 2 1 Instituto de Ecorregiones Andinas (INECOA)-Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Avenida Bolivia 1239, San Salvador de Jujuy, Jujuy,Argentina. 2 Laboratorio de Microbiología y Sanidad Apícola, Facultad de Ciencias Agrarias, Universidad Nacional de Jujuy, Alberdi 47, San Salvador de Jujuy, Jujuy, Argentina; [email protected] * Corresponding author E-mail: [email protected] ABSTRACT Ants are frequent visitors to Apis mellifera L. hives. Certain species may cause serious loss of productivity to apiculture. The objective of this study was to describe the diversity and abundance of ants both outside and inside the hives of Apis mellifera L. in Valles Templados, Jujuy province, Argentine. Ants were collected by hand and pitfall traps in the Spring and Summer season of 2016, 2017 and 2018, in apiaries found at three different localities of Valles Templados: Río Blanco, Tilquiza and Severino. Pitfall trapping resulted in 824 specimens belonging to 10 species in Severino, 208 individuals of 9 species in Tilquiza, and 289 individuals of 2 species in Río Blanco. The most common ant species found were: Pheidole sp. (56.07%), Linepithema humile (94.81%), and Solenopsis sp. (56.73%) in Severino, Rio Blanco and Tilquiza, respectively. -

Maunakea Invertebrate Threats, Identification, Collection, and Processing Guide
Office of Mauna Kea Management Invasive Species Plan Standard Operating Procedure C Maunakea Invertebrate Threats, Identification, Collection, and Processing Guide Version 1.0, 2/10/2015, Jessica Kirkpatrick & Fritz Klasner Table of Contents 1. Purpose and Scope .................................................................................................................................... 1 Invertebrate Threats ................................................................................................................................. 2 2. List of Common and Targeted Invertebrates ............................................................................................ 2 3. Identification of Common and Targeted Invertebrates ............................................................................ 4 4. Arthropod Collection............................................................................................................................... 14 Collection Supplies .................................................................................................................................. 14 Collecting Potential Threats .................................................................................................................... 14 Hand Search & Haphazard Collections.................................................................................................... 14 Flying Arthropods ............................................................................................................................... -

The Ant Genus Linepithema (Formicidae: Dolichoderinae) in Colombia
Zootaxa 4208 (5): 446–458 ISSN 1175-5326 (print edition) http://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2016 Magnolia Press ISSN 1175-5334 (online edition) http://doi.org/10.11646/zootaxa.4208.5.3 http://zoobank.org/urn:lsid:zoobank.org:pub:69728BA3-6562-43A0-9A8F-6DCF61817BD1 The ant genus Linepithema (Formicidae: Dolichoderinae) in Colombia MAYRON ESCÁRRAGA1, 2,3 & ROBERTO J. GUERRERO1 1Grupo Insectos Neotropicales, Facultad de Ciencias Básicas, Universidad del Magdalena, Carrera 32 # 22-08, Santa Marta, Colom- bia 2Programa de Pós graduação em Entomologia, Universidade Federal do Paraná, CEP: 81531-980 - Curitiba, Paraná, Brasil 3Corresponding author. E-mail: [email protected] Abstract The species of Linepithema Mayr in Colombia are reviewed from collection material. The genus Linepithema comprises 20 species, of which the following are reported in Colombia: L. angulatum (Emery), L. dispertitum (Forel), L. gallardoi (Brèthes), L. humile (Mayr), L. iniquum (Mayr), L. neotropicum Wild, L. piliferum (Mayr), L. tsachila Wild, and a new species, Linepithema hirsutum sp. nov. Linepithema dispertitum is a new record for Colombia and South America. A tax- onomic key (English and Spanish) and distribution maps to the species of Linepithema in Colombia are provided. Key words: geographical distribution, Neotropical region, new record, new species, taxonomic keys Resumen Se revisaron las especies de Linepithema Mayr en Colombia a partir de material de colecciones. El género Linepithema comprende 20 especies, de las cuales en Colombia se reportan las siguientes: L. angulatum (Emery), L. dispertitum (Forel), L. gallardoi (Brèthes), L. humile (Mayr), L. iniquum (Mayr), L. neotropicum Wild, L. piliferum (Mayr), L. -

Bioinvasions in a Changing World: a Resource on Invasive Species-Climate Change Interactions for Conservation and Natural Resource Management
Bioinvasions in a Changing World: A Resource on Invasive Species-Climate Change Interactions for Conservation and Natural Resource Management December 2014 Prepared for The Aquatic Nuisance Species Task Force (ANSTF) and The National Invasive Species Council (NISC) By the Ad Hoc Working Group on Invasive Species and Climate Change The Ad Hoc Working Group on Invasive Species and Climate Change Co-chairs: Stanley W. Burgiel (NISC) Thomas Hall (USDA Animal and Plant Health Inspection Service) Working group members: Noah Adams (U.S. Geological Survey) Denny Lassuy (North Slope Science Initiative) Kevin Anderson (Puget Sound Partnership) Louanne McMartin (U.S. Fish and Wildlife Elizabeth Bella (U.S. Fish and Wildlife Service) Service) Britta Bierwagen (Environmental Protection Phil Moy (University of Wisconsin Sea Grant Agency) Institute) Maria Boroja (U.S. Fish and Wildlife Service) Adrianna Muir (Department of Interior) Elizabeth Brusati (California Invasive Plant Richard Nelson (U.S. Fish and Wildlife Service) Council) Judith Pederson (MIT Sea Grant College Jeff Burgett (U.S. Fish and Wildlife Service) Program) LaDonna Carlisle (Bureau of Indian Affairs) Glenn Plumb (National Park Service) Ruark Cleary (Florida Fish and Wildlife Jim Quinn (University of California-Davis) Conservation Commission) Jennifer Resnick (National Park Service) Dave Cleland (U.S. Forest Service) Dolores Savignano (U.S. Fish and Wildlife John Darling (Environmental Protection Agency) Service) Robyn Draheim (U.S. Fish and Wildlife Service) Nick Schmal (U.S. Forest Service) Darryl Forest (Department of Defense) Linda Shaw (National Oceanic and Atmospheric Sara Grise-Stahlman (Pennsylvania Sea Grant) Administration) Sean Hart (Bureau of Indian Affairs) Bruce Stein (National Wildlife Federation) Keith Hatch (Bureau of Indian Affairs) Genelle Winter (Metlakatla Indian Community) Paul Heimowitz (U.S.