Exploring Archaeal and Bacterial Diversity and Co-Occurrence in Lake Geneva Jade Ezzedine, Yves Desdevises, Stéphan Jacquet
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genome of Nanoarchaeum Equitans: Insights Into Early Archaeal Evolution and Derived Parasitism
The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism Elizabeth Waters†‡, Michael J. Hohn§, Ivan Ahel¶, David E. Graham††, Mark D. Adams‡‡, Mary Barnstead‡‡, Karen Y. Beeson‡‡, Lisa Bibbs†, Randall Bolanos‡‡, Martin Keller†, Keith Kretz†, Xiaoying Lin‡‡, Eric Mathur†, Jingwei Ni‡‡, Mircea Podar†, Toby Richardson†, Granger G. Sutton‡‡, Melvin Simon†, Dieter So¨ ll¶§§¶¶, Karl O. Stetter†§¶¶, Jay M. Short†, and Michiel Noordewier†¶¶ †Diversa Corporation, 4955 Directors Place, San Diego, CA 92121; ‡Department of Biology, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182; §Lehrstuhl fu¨r Mikrobiologie und Archaeenzentrum, Universita¨t Regensburg, Universita¨tsstrasse 31, D-93053 Regensburg, Germany; ‡‡Celera Genomics Rockville, 45 West Gude Drive, Rockville, MD 20850; Departments of ¶Molecular Biophysics and Biochemistry and §§Chemistry, Yale University, New Haven, CT 06520-8114; and ʈDepartment of Biochemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061 Communicated by Carl R. Woese, University of Illinois at Urbana–Champaign, Urbana, IL, August 21, 2003 (received for review July 22, 2003) The hyperthermophile Nanoarchaeum equitans is an obligate sym- (6–8). Genomic DNA was either digested with restriction en- biont growing in coculture with the crenarchaeon Ignicoccus. zymes or sheared to provide clonable fragments. Two plasmid Ribosomal protein and rRNA-based phylogenies place its branching libraries were made by subcloning randomly sheared fragments point early in the archaeal lineage, representing the new archaeal of this DNA into a high-copy number vector (Ϸ2.8 kbp library) kingdom Nanoarchaeota. The N. equitans genome (490,885 base or low-copy number vector (Ϸ6.3 kbp library). DNA sequence pairs) encodes the machinery for information processing and was obtained from both ends of plasmid inserts to create repair, but lacks genes for lipid, cofactor, amino acid, or nucleotide ‘‘mate-pairs,’’ pairs of reads from single clones that should be biosyntheses. -

Bacterial Diversity in the Surface Sediments of the Hypoxic Zone Near
ORIGINAL RESEARCH Bacterial diversity in the surface sediments of the hypoxic zone near the Changjiang Estuary and in the East China Sea Qi Ye, Ying Wu, Zhuoyi Zhu, Xiaona Wang, Zhongqiao Li & Jing Zhang State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai 200062, China Keywords Abstract Bacteria, Changjiang Estuary, hypoxia, Miseq Illumina sequencing, sediment Changjiang (Yangtze River) Estuary has experienced severe hypoxia since the 1950s. In order to investigate potential ecological functions of key microorgan- Correspondence isms in relation to hypoxia, we performed 16S rRNA-based Illumina Miseq Qi Ye, East China Normal University, State Key sequencing to explore the bacterial diversity in the surface sediments of the Laboratory of Estuarine and Coastal Research, hypoxic zone near the Changjiang Estuary and in the East China Sea (ECS). 3663 North Zhongshan Road, SKLEC Building, Room 419, Shanghai 200062, China. The results showed that numerous Proteobacteria-affiliated sequences in the sedi- Tel: 86-021-52124974; ments of the inner continental shelf were related to both sulfate-reducing and Fax: 86-021- 62546441; sulfur-oxidizing bacteria, suggesting an active sulfur cycle in this area. Many E-mail: [email protected] sequences retrieved from the hypoxic zone were also related to Planctomycetes from two marine upwelling systems, which may be involved in the initial break- Funding Information down of sulfated heteropolysaccharides. Bacteroidetes, which is expected to degrade This study was funded by the Shanghai Pujiang high-molecular-weight organic matter, was abundant in all the studied stations Talent Program (12PJ1403100), the National except for station A8, which was the deepest and possessed the largest grain Natural Science Foundation of China (41276081), the Key Project of Chinese size. -

University of Oklahoma Graduate College Development of High
UNIVERSITY OF OKLAHOMA GRADUATE COLLEGE DEVELOPMENT OF HIGH-THROUGHPUT EXPERIMENTAL AND COMPUTATIONAL TECHNOLOGIES FOR ANALYZING MICROBIAL FUNCTIONS AND INTERACTIONS IN ENVIRONMENTAL METAGENOMES A DISSERTATION SUBMITTED TO THE GRADUATE FACULTY in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY By ZHOU SHI Norman, Oklahoma 2017 DEVELOPMENT OF HIGH-THROUGHPUT EXPERIMENTAL AND COMPUTATIONAL TECHNOLOGIES FOR ANALYZING MICROBIAL FUNCTIONS AND INTERACTIONS IN ENVIRONMENTAL METAGENOMES A DISSERTATION APPROVED FOR THE DEPARTMENT OF MICROBIOLOGY AND PLANT BIOLOGY BY ______________________________ Dr. Lee Krumholz, Chair ______________________________ Dr. Meijun Zhu ______________________________ Dr. Sridhar Radhakrishnan ______________________________ Dr. Yiqi Luo ______________________________ Dr. Jizhong Zhou © Copyright by ZHOU SHI 2017 All Rights Reserved. Dedication I dedicate this dissertation to my wife, Mengting Yuan, whom I met and married during the time period of this work and whose love, faith and support is the greatest gift that I can ever imagine in my life; to my son, Andrew Shi, who is always curious, energetic and wonderful, and is my lasting source of enlightenment and true happiness; to my parents, Yiping Shi and Aiping Zeng, whose selfless love and care transcended the distance and time, and carried me whenever I need; to my aunt, Cindy Shi, whose unwavering support and encouragement instilled in me the spirit of striving for excellence; to my friend, Su Xu, who boarded on the same plane with me to the U.S. seven years ago and then has witnessed all parts of this work as my best friend, and Dian Zou, who passed away in an accident during the time period of this work and is a friend I will forever miss. -

Yu-Chen Ling and John W. Moreau
Microbial Distribution and Activity in a Coastal Acid Sulfate Soil System Introduction: Bioremediation in Yu-Chen Ling and John W. Moreau coastal acid sulfate soil systems Method A Coastal acid sulfate soil (CASS) systems were School of Earth Sciences, University of Melbourne, Melbourne, VIC 3010, Australia formed when people drained the coastal area Microbial distribution controlled by environmental parameters Microbial activity showed two patterns exposing the soil to the air. Drainage makes iron Microbial structures can be grouped into three zones based on the highest similarity between samples (Fig. 4). Abundant populations, such as Deltaproteobacteria, kept constant activity across tidal cycling, whereas rare sulfides oxidize and release acidity to the These three zones were consistent with their geological background (Fig. 5). Zone 1: Organic horizon, had the populations changed activity response to environmental variations. Activity = cDNA/DNA environment, low pH pore water further dissolved lowest pH value. Zone 2: surface tidal zone, was influenced the most by tidal activity. Zone 3: Sulfuric zone, Abundant populations: the heavy metals. The acidity and toxic metals then Method A Deltaproteobacteria Deltaproteobacteria this area got neutralized the most. contaminate coastal and nearby ecosystems and Method B 1.5 cause environmental problems, such as fish kills, 1.5 decreased rice yields, release of greenhouse gases, Chloroflexi and construction damage. In Australia, there is Gammaproteobacteria Gammaproteobacteria about a $10 billion “legacy” from acid sulfate soils, Chloroflexi even though Australia is only occupied by around 1.0 1.0 Cyanobacteria,@ Acidobacteria Acidobacteria Alphaproteobacteria 18% of the global acid sulfate soils. Chloroplast Zetaproteobacteria Rare populations: Alphaproteobacteria Method A log(RNA(%)+1) Zetaproteobacteria log(RNA(%)+1) Method C Method B 0.5 0.5 Cyanobacteria,@ Bacteroidetes Chloroplast Firmicutes Firmicutes Bacteroidetes Planctomycetes Planctomycetes Ac8nobacteria Fig. -

Cultivating the Macroalgal Holobiont: Effects of Integrated Multi-Trophic Aquaculture on the Microbiome of Ulva Rigida (Chlorophyta)
fmars-07-00052 February 10, 2020 Time: 15:0 # 1 ORIGINAL RESEARCH published: 12 February 2020 doi: 10.3389/fmars.2020.00052 Cultivating the Macroalgal Holobiont: Effects of Integrated Multi-Trophic Aquaculture on the Microbiome of Ulva rigida (Chlorophyta) Gianmaria Califano1, Michiel Kwantes1, Maria Helena Abreu2, Rodrigo Costa3,4,5 and Thomas Wichard1,6* 1 Institute for Inorganic and Analytical Chemistry, Friedrich Schiller University Jena, Jena, Germany, 2 ALGAplus Lda, Ílhavo, Portugal, 3 Institute for Bioengineering and Biosciences, Instituto Superior Técnico, Universidade de Lisboa, Lisbon, Portugal, 4 Centre of Marine Sciences, University of Algarve, Faro, Portugal, 5 Lawrence Berkeley National Laboratory, U.S. Department of Energy Joint Genome Institute, University of California, Berkeley, Berkeley, CA, United States, 6 Jena School for Microbial Communication, Jena, Germany Ulva is a ubiquitous macroalgal genus of commercial interest. Integrated Multi-Trophic Aquaculture (IMTA) systems promise large-scale production of macroalgae due to Edited by: their high productivity and environmental sustainability. Complex host–microbiome Bernardo Antonio Perez Da Gama, interactions play a decisive role in macroalgal development, especially in Ulva spp. Universidade Federal Fluminense, due to algal growth- and morphogenesis-promoting factors released by associated Brazil bacteria. However, our current understanding of the microbial community assembly and Reviewed by: Alejandro H. Buschmann, structure in cultivated macroalgae is scant. We aimed to determine (i) to what extent University of Los Lagos, Chile IMTA settings influence the microbiome associated with U. rigida and its rearing water, (ii) Henrique Fragoso Santos, to explore the dynamics of beneficial microbes to algal growth and development under Universidade Federal Fluminense, Brazil IMTA settings, and (iii) to improve current knowledge of host–microbiome interactions. -

Insights Into Archaeal Evolution and Symbiosis from the Genomes of a Nanoarchaeon and Its Inferred Crenarchaeal Host from Obsidian Pool, Yellowstone National Park
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Microbiology Publications and Other Works Microbiology 4-22-2013 Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park Mircea Podar University of Tennessee - Knoxville, [email protected] Kira S. Makarova National Institutes of Health David E. Graham University of Tennessee - Knoxville, [email protected] Yuri I. Wolf National Institutes of Health Eugene V. Koonin National Institutes of Health See next page for additional authors Follow this and additional works at: https://trace.tennessee.edu/utk_micrpubs Part of the Microbiology Commons Recommended Citation Biology Direct 2013, 8:9 doi:10.1186/1745-6150-8-9 This Article is brought to you for free and open access by the Microbiology at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Microbiology Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Authors Mircea Podar, Kira S. Makarova, David E. Graham, Yuri I. Wolf, Eugene V. Koonin, and Anna-Louise Reysenbach This article is available at TRACE: Tennessee Research and Creative Exchange: https://trace.tennessee.edu/ utk_micrpubs/44 Podar et al. Biology Direct 2013, 8:9 http://www.biology-direct.com/content/8/1/9 RESEARCH Open Access Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park Mircea Podar1,2*, Kira S Makarova3, David E Graham1,2, Yuri I Wolf3, Eugene V Koonin3 and Anna-Louise Reysenbach4 Abstract Background: A single cultured marine organism, Nanoarchaeum equitans, represents the Nanoarchaeota branch of symbiotic Archaea, with a highly reduced genome and unusual features such as multiple split genes. -

The Bacterial Communities of Sand-Like Surface Soils of the San Rafael Swell (Utah, USA) and the Desert of Maine (USA) Yang Wang
The bacterial communities of sand-like surface soils of the San Rafael Swell (Utah, USA) and the Desert of Maine (USA) Yang Wang To cite this version: Yang Wang. The bacterial communities of sand-like surface soils of the San Rafael Swell (Utah, USA) and the Desert of Maine (USA). Agricultural sciences. Université Paris-Saclay, 2015. English. NNT : 2015SACLS120. tel-01261518 HAL Id: tel-01261518 https://tel.archives-ouvertes.fr/tel-01261518 Submitted on 25 Jan 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2015SACLS120 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N°577 Structure et Dynamique des Systèmes Vivants Spécialité de doctorat : Sciences de la Vie et de la Santé Par Mme Yang WANG The bacterial communities of sand-like surface soils of the San Rafael Swell (Utah, USA) and the Desert of Maine (USA) Thèse présentée et soutenue à Orsay, le 23 Novembre 2015 Composition du Jury : Mme. Marie-Claire Lett , Professeure, Université Strasbourg, Rapporteur Mme. Corinne Cassier-Chauvat , Directeur de Recherche, CEA, Rapporteur M. Armel Guyonvarch, Professeur, Université Paris-Sud, Président du Jury M. -

Lifestyle Preferences Drive the Structure and Diversity of Bacterial and Archaeal Communities in a Small Riverine Reservoir
www.nature.com/scientificreports OPEN Lifestyle preferences drive the structure and diversity of bacterial and archaeal communities in a small riverine reservoir Carles Borrego1,2, Sergi Sabater1,3* & Lorenzo Proia1,4 Spatial heterogeneity along river networks is interrupted by dams, afecting the transport, processing, and storage of organic matter, as well as the distribution of biota. We here investigated the structure of planktonic (free-living, FL), particle-attached (PA) and sediment-associated (SD) bacterial and archaeal communities within a small reservoir. We combined targeted-amplicon sequencing of bacterial and archaeal 16S rRNA genes in the DNA and RNA community fractions from FL, PA and SD, followed by imputed functional metagenomics, in order to unveil diferences in their potential metabolic capabilities within the reservoir (tail, mid, and dam sections) and lifestyles (FL, PA, SD). Both bacterial and archaeal communities were structured according to their life-style preferences rather than to their location in the reservoir. Bacterial communities were richer and more diverse when attached to particles or inhabiting the sediment, while Archaea showed an opposing trend. Diferences between PA and FL bacterial communities were consistent at functional level, the PA community showing higher potential capacity to degrade complex carbohydrates, aromatic compounds, and proteinaceous materials. Our results stressed that particle-attached prokaryotes were phylogenetically and metabolically distinct from their free-living counterparts, and that performed as hotspots for organic matter processing within the small reservoir. Spatial heterogeneity of river networks results from the sequence of lotic segments—which promote the transport and quick transformation of materials—and lentic segments such as large pools and wetlands—which mostly contribute to the process and storage of organic matter 1,2. -

Archaea and the Origin of Eukaryotes
REVIEWS Archaea and the origin of eukaryotes Laura Eme, Anja Spang, Jonathan Lombard, Courtney W. Stairs and Thijs J. G. Ettema Abstract | Woese and Fox’s 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor. Sister groups Two descendants that split The pioneering work by Carl Woese and colleagues In this Review, we discuss how culture- independent from the same node; the revealed that all cellular life could be divided into three genomics has transformed our understanding of descendants are each other’s major evolutionary lines (also called domains): the archaeal diversity and how this has influenced our closest relative. -

1 Characterization of Sulfur Metabolizing Microbes in a Cold Saline Microbial Mat of the Canadian High Arctic Raven Comery Mast
Characterization of sulfur metabolizing microbes in a cold saline microbial mat of the Canadian High Arctic Raven Comery Master of Science Department of Natural Resource Sciences Unit: Microbiology McGill University, Montreal July 2015 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master in Science © Raven Comery 2015 1 Abstract/Résumé The Gypsum Hill (GH) spring system is located on Axel Heiberg Island of the High Arctic, perennially discharging cold hypersaline water rich in sulfur compounds. Microbial mats are found adjacent to channels of the GH springs. This thesis is the first detailed analysis of the Gypsum Hill spring microbial mats and their microbial diversity. Physicochemical analyses of the water saturating the GH spring microbial mat show that in summer it is cold (9°C), hypersaline (5.6%), and contains sulfide (0-10 ppm) and thiosulfate (>50 ppm). Pyrosequencing analyses were carried out on both 16S rRNA transcripts (i.e. cDNA) and genes (i.e. DNA) to investigate the mat’s community composition, diversity, and putatively active members. In order to investigate the sulfate reducing community in detail, the sulfite reductase gene and its transcript were also sequenced. Finally, enrichment cultures for sulfate/sulfur reducing bacteria were set up and monitored for sulfide production at cold temperatures. Overall, sulfur metabolism was found to be an important component of the GH microbial mat system, particularly the active fraction, as 49% of DNA and 77% of cDNA from bacterial 16S rRNA gene libraries were classified as taxa capable of the reduction or oxidation of sulfur compounds. -
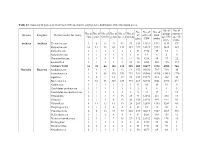
Domain Kingdom Phylum (Order for Virus) No. of Class No. of Order No. of Family No. of Genus No. of Species No. of Strains No. O
Table S2. Summary of probes on GeoChip 5.0M based on the phylogenetic distribution of the functional genes. No. of No. of No. No. of No. of No. of No. of No. of No. of No. of No. of group- sequence Domain Kingdom Phylum (order for virus) of covered total class order family genus species strains specific -specific gene CDS probe probe probe Archaea Archaea Crenarchaeota 2 6 9 22 43 57 142 13921 1312 1133 179 Euryarchaeota 10 12 23 69 133 187 229 33323 3992 3625 367 Korarchaeota 1 1 1 1 1 1 33 3718 35 33 2 Nanoarchaeota 1 1 1 1 1 1 6 12 6 6 0 Thaumarchaeota 1 4 5 7 9 14 46 5218 95 73 22 unclassified 1 1 1 1 1 25 46 1885 288 115 173 Archaea Total 16 25 40 101 188 285 502 58077 5728 4985 743 Bacteria Bacteria Acidobacteria 5 4 4 8 13 18 155 20180 745 715 30 Actinobacteria 1 8 48 120 351 753 381 80060 15394 13618 1776 Aquificae 1 2 4 12 17 22 127 19379 533 498 35 Bacteroidetes 7 10 23 107 239 394 267 58970 9661 8790 871 Caldiserica 1 1 1 1 1 1 13 38 19 19 0 Candidatus poribacteria 1 1 1 1 1 1 2 5 4 1 3 Candidatus saccharibacteria 1 1 1 1 1 3 9 22 17 4 13 Chlamydiae 2 1 4 7 15 23 74 12652 241 230 11 Chlorobi 1 1 1 6 15 20 138 21932 995 944 51 Chloroflexi 8 11 12 14 19 28 209 32839 1353 1289 64 Chrysiogenetes 1 1 1 2 2 2 33 81 41 40 1 Cyanobacteria 2 8 12 57 97 203 237 46219 4727 4209 518 Deferribacteres 1 1 1 4 4 4 81 2583 165 154 11 Deinococcus-thermus 2 3 4 7 18 27 172 21352 1026 978 48 Dictyoglomi 1 1 1 1 2 2 51 9447 99 98 1 Elusimicrobia 2 2 2 2 2 2 34 5713 50 49 1 Fibrobacteres 2 1 1 1 1 2 50 6671 69 64 5 Firmicutes 6 10 42 176 593 1556 -

Concerted Gene Recruitment in Early Plant Evolution Jinling Huang* and J Peter Gogarten†
Open Access Research2008HuangVolume and 9, Issue Gogarten 7, Article R109 Concerted gene recruitment in early plant evolution Jinling Huang* and J Peter Gogarten† Addresses: *Department of Biology, Howell Science Complex, East Carolina University, Greenville, NC 27858, USA. †Department of Molecular and Cell Biology, University of Connecticut, 91 North Eagleville Road, Storrs, CT 06269, USA. Correspondence: Jinling Huang. Email: [email protected] Published: 8 July 2008 Received: 30 April 2008 Revised: 24 June 2008 Genome Biology 2008, 9:R109 (doi:10.1186/gb-2008-9-7-r109) Accepted: 8 July 2008 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2008/9/7/R109 © 2008 Huang and Gogarten; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Eukaryotic<p>Analysesto the split ofhorizontal ofred the algae red gene andalgal transfergreen <it>Cyanidioschyzon</it> plants.</p> genome identified 37 genes that were acquired from non-organellar sources prior Abstract Background: Horizontal gene transfer occurs frequently in prokaryotes and unicellular eukaryotes. Anciently acquired genes, if retained among descendants, might significantly affect the long-term evolution of the recipient lineage. However, no systematic studies on the scope of anciently acquired genes and their impact on macroevolution are currently available in eukaryotes. Results: Analyses of the genome of the red alga Cyanidioschyzon identified 37 genes that were acquired from non-organellar sources prior to the split of red algae and green plants.