Identification and Characterization of the Arabidopsis Gene Encoding The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Medications in Pregnant and Nursing Mothers
Medications in Pregnant and Nursing Mothers NADINE M. GIRGIS, OD, FAAO ASSISTANT PROFESSOR YIN C. TEA, OD, FAAO CHIEF, PEDIATRICS AND BINOCULAR VISION ASSISTANT PROFESSOR Gestation age vs fetal age Gestation age-sperm penetrates the egg and zygote is formed Zygote (fertilized egg) travels from fallopian tube to uterus During this time, egg divides into cells - called a morula Continued dividing and morula - called a blastocyst - embeds in the uterus anywhere from 6-12 days after conception This begins the embryonic stage and fetal age begins Fetal development-1st trimester Gestation age week 3-fetal age week 1: a lot of basic growth Brain, spinal cord, heart, GI tract begin development 1st trimester Gestation age-week 4 and 5: embryo ¼ inch long Arm and leg buds, ears, eyes forming Placenta forming and producing hormones Heart is beating at a steady rhythm Movement of rudimentary blood through blood vessels 1st trimester Gestation age week 6: embryo is ½ in length Lungs, jaw, nose, plate formation, hands and feet Hand and feet buds have webbed structures Brain forming into complex parts 1st trimester Gestation age week 7: weighs less than an aspirin All essential organs have begun to form Hair, nail follicles, eyelids and tongue starting to form Trunk begins to straighten out 1st trimester Gestation age week 8: 1 in long, size of a bean All parts of adult are now present in the embryo Bones beginning to form Muscles begin to contract Facial features, including eyelids more developed Gestation age weeks 9-13: 3 in and weighs -

Preferred Drug List
October 2021 Preferred Drug List The Preferred Drug List, administered by CVS Caremark® on behalf of Siemens, is a guide within select therapeutic categories for clients, plan members and health care providers. Generics should be considered the first line of prescribing. If there is no generic available, there may be more than one brand-name medicine to treat a condition. These preferred brand-name medicines are listed to help identify products that are clinically appropriate and cost-effective. Generics listed in therapeutic categories are for representational purposes only. This is not an all-inclusive list. This list represents brand products in CAPS, branded generics in upper- and lowercase Italics, and generic products in lowercase italics. PLAN MEMBER HEALTH CARE PROVIDER Your benefit plan provides you with a prescription benefit program Your patient is covered under a prescription benefit plan administered administered by CVS Caremark. Ask your doctor to consider by CVS Caremark. As a way to help manage health care costs, prescribing, when medically appropriate, a preferred medicine from authorize generic substitution whenever possible. If you believe a this list. Take this list along when you or a covered family member brand-name product is necessary, consider prescribing a brand name sees a doctor. on this list. Please note: Please note: • Your specific prescription benefit plan design may not cover • Generics should be considered the first line of prescribing. certain products or categories, regardless of their appearance in • This drug list represents a summary of prescription coverage. It is this document. Products recently approved by the U.S. Food and not all-inclusive and does not guarantee coverage. -

Specific Pattern of Gene Expression During Induction of Mouse Erythroleukemia Cells
Downloaded from genesdev.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Specific pattern of gene expression during induction of mouse erythroleukemia cells Peter J. Fraser and Peter J. Curtis The Wistar Institute of Anatomy and Biology, Philadelphia, Pennsylvania 19104 USA We have studied the expression of several characterized genes during induction of mouse erythroleukemia (MEL) cells with dimethyl sulfoxide (DMSO) and have observed a specific pattern of changes in transcriptional activity and steady-state RNA levels associated with erythroid differentiation. During induction there is a gradual, steady decrease in total transcriptional activity and RNA content per cell, which by day 3 of DMSO treatment amounts to less than 50% of the level in the uninduced cell. During this time we observe increases in transcriptional activity for 5-aminolevulinic acid synthase, carbonic anhydrase form II, and band 3 coordinate with the large increase in [3-globin gene transcription. The results also demonstrate an early decrease in transcription for carbonic anhydrase form I, which precedes decreases in transcription for glyceraldehyde phosphate dehydrogenase and rRNA genes. Changes in steady-state RNA levels reflected changes in transcriptional activity during induction except for carbonic anhydrase II mRNA. These results represent the first report characterizing the regulated expression at transcriptional and posttranscriptional levels of several known genes that are characteristically expressed in the erythrocyte. The results demonstrate that coordinate gene expression in erythroid differentiation occurs primarily at the level of transcription. [Key Words: Coordinated gene regulation; transcriptional activity; erythroid differentiation] Received June 25, 1987; revised version accepted August 14, 1987. Adult erythroid differentiation requires activation and/ dinately such that they may assemble to form the char- or modulation of many genes whose expression is regu- acteristic erythrocyte cytoskeleton. -
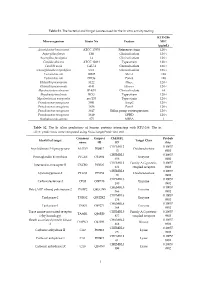
Table S1. the Bacterial and Fungal Isolates Used for the in Vitro Activity Testing
Table S1. The bacterial and fungal isolates used for the in vitro activity testing. KTU-286 Microorganism Strain No Feature MIC (µg/mL) Acinetobacter baummanii ATCC 17978 Reference strain 128 < Aspergillus flavus 12B Clinical isolate 128 < Aspergillus fumigatus 14 Clinical isolate 128 < Candida albicans ATCC 10231 Type strain 128 < Candida auris CAU-1 Clinical isolate 128 < Cuninghamella corymbifera CC1 Clinical isolate 128 < Escherichia coli 10025 Mcr-1 128 Escherichia coli DH5a Pan-S 128 Klebsiella pneumoniae 3122 blaKPC 128 < Klebsiella pneumoniae 4141 blaNDM-1 128 < Mycobacterium abscesus IP-K01 Clinical isolate 64 Mycobacterium bovis BCG Type strain 128 < Mycobacterium smegmatis mc2155 Type strain 128 < Pseudomonas aeruginosa 3691 AmpC 128 < Pseudomonas aeruginosa 3656 Pan-S 128 < Pseudomonas aeruginosa 3647 Efflux pump overexpression 128 < Pseudomonas aeruginosa 3619 OPRD 128 < Staphylococcus aureus 875 MRSA 1 Table S2. The In silico predictions of human proteins interacting with KTU-286. The in silico predictions were computed using SwissTargetPrediction tool. Common Uniprot ChEMBL Probab Identified target Target Class name ID ID ility CHEMBL2 0.10057 Arachidonate 5-lipoxygenase ALOX5 P09917 Oxidoreductase 15 8902 CHEMBL5 0.10057 Prostaglandin E synthase PTGES O14684 Enzyme 658 8902 CHEMBL2 Family A G protein- 0.10057 Interleukin-8 receptor B CXCR2 P25025 434 coupled receptor 8902 CHEMBL2 0.10057 Cyclooxygenase-2 PTGS2 P35354 Oxidoreductase 30 8902 CHEMBL3 0.10057 Carboxylesterase 2 CES2 O00748 Enzyme 180 8902 CHEMBL5 0.10057 Poly -

Targeting Mycobacterium Tuberculosis Proteins: Structure and Function Studies of Five Essential Proteins
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 411 Targeting Mycobacterium tuberculosis Proteins: Structure and Function Studies of Five Essential Proteins ADRIAN SUAREZ COVARRUBIAS ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 UPPSALA ISBN 978-91-554-7134-7 2008 urn:nbn:se:uu:diva-8580 !! ""# !$"" % & % % '& &( )& * + &( ,- ( ""#( ) ' $ , , % + ' ( ( !!( .! ( ( /,0 12#31!34432!32( )& & & % - & &- % % 56 ( )& & % & % & % %% & ( )& % & 5 6 7!# 74## & (" !(2 8 ( 74## 7!# 93 & & * * %% % 3 - ( 7!14 & & *& && : & & (4 8 *&& % & & & ( & &; & %% ( < % & % & 5 6( )& & % - & * & % ( 7".. % ; * 3% & 93& 3 ' & *&& % &( 3 *& ( 44= % & ; * % ( 722# % & &3 & !(# 8 ( , & % & % & * ( &* ( & & & , // 7".. 7!# 7!14 74## 722# >3 & ! " # " #$ %&'" " ()*%+,- " ? ,- ""# /,,0 !.4!3.! /,0 12#31!34432!32 $ $$$ 3#4#" 5& $@@ (;(@ A B $ $$$ 3#4#"6 List of publications This thesis consists of a comprehensive summary based on the following papers. In the text, the papers will be referred to by their roman numerals. I Covarrubias, A. S., Larsson, A. M., Högbom, M., Lindberg, J., Bergfors, T., Björkelid, C., Mowbray, S. L., Unge, T., and Jones, T. A. (2005). Structure and -

The Green Synthesis and Molecular Docking of Novel N-Substituted
Bioorganic Chemistry 90 (2019) 103096 Contents lists available at ScienceDirect Bioorganic Chemistry journal homepage: www.elsevier.com/locate/bioorg The green synthesis and molecular docking of novel N-substituted T rhodanines as effective inhibitors for carbonic anhydrase and acetylcholinesterase enzymes ⁎ ⁎ Sinan Bayindira, , Cuneyt Caglayanb, Muhammet Karamanc, İlhami Gülcind, a Department of Chemistry, Faculty of Sciences and Arts, Bingol University, 12000-Bingöl, Turkey b Department of Biochemistry, Faculty of Veterinary Medicine, Bingol University, 12000-Bingöl, Turkey c Department of Molecular Biology and Genetics, Faculty of Arts and Science, Kilis 7 Aralik University, 79000-Kilis, Turkey d Department of Chemistry, Faculty of Sciences, Atatürk University, 25240-Erzurum, Turkey ARTICLE INFO ABSTRACT Keywords: Recently, inhibition effects of enzymes such as acetylcholinesterase (AChE) and carbonic anhydrase (CA)has Rhodanine appeared as a promising approach for pharmacological intervention in a variety of disorders such as epilepsy, Aza-ylides Alzheimer’s disease and obesity. For this purpose, novel N-substituted rhodanine derivatives (RhAs) were syn- Acetylcholinesterase thesized by a green synthetic approach over one-pot reaction. Following synthesis the novel compounds, RhAs Carbonic anhydrase derivatives were tested against AChE and cytosolic carbonic anhydrase I, and II (hCAs I, and II) isoforms. As a Enzyme inhibition result of this study, inhibition constant (Ki) were found in the range of 66.35 ± 8.35 to 141.92 ± 12.63 nM for Molecular docking AChE, 43.55 ± 14.20 to 89.44 ± 24.77 nM for hCA I, and 16.97 ± 1.42 to 64.57 ± 13.27 nM for hCA II, respectively. Binding energies were calculated with docking studies as −5.969, −5.981, and −9.121 kcal/mol for hCA I, hCA II, and AChE, respectively. -

Regulation of Pancreatic Islet Gene Expression in Mouse Islets by Pregnancy
265 Regulation of pancreatic islet gene expression in mouse islets by pregnancy B T Layden, V Durai, M V Newman, A M Marinelarena, C W Ahn, G Feng1, S Lin1, X Zhang2, D B Kaufman2, N Jafari3, G L Sørensen4 and W L Lowe Jr Division of Endocrinology, Metabolism and Molecular Medicine, Department of Medicine, Northwestern University Feinberg School of Medicine, 303 East Chicago Avenue, Tarry 15, Chicago, Illinois 60611, USA 1Northwestern University Biomedical Informatics Center, 2Division of Transplantation Surgery, Department of Surgery and 3Genomics Core, Center for Genetic Medicine, Northwestern University, Chicago, Illinois 60611, USA 4Medical Biotechnology Center, University of Southern Denmark, DK-5000 Odense C, Denmark (Correspondence should be addressed to W L Lowe Jr; Email: [email protected]) Abstract Pancreatic b cells adapt to pregnancy-induced insulin were confirmed in murine islets. Cytokine-induced resistance by unclear mechanisms. This study sought to expression of SP-D in islets was also demonstrated, suggesting identify genes involved in b cell adaptation during pregnancy. a possible role as an anti-inflammatory molecule. Comple- To examine changes in global RNA expression during menting these studies, an expression array was performed to pregnancy, murine islets were isolated at a time point of define pregnancy-induced changes in expression of GPCRs increased b cell proliferation (E13.5), and RNA levels were that are known to impact islet cell function and proliferation. determined by two different assays (global gene expression This assay, the results of which were confirmed using real- array and G-protein-coupled receptor (GPCR) array). time reverse transcription-PCR assays, demonstrated that free Follow-up studies confirmed the findings for select genes. -

Dithiocarbamates with Potent Inhibitory Activity Against the Saccharomyces Cerevisiae Β-Carbonic Anhydrase
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Florence Research Journal of Enzyme Inhibition and Medicinal Chemistry ISSN: 1475-6366 (Print) 1475-6374 (Online) Journal homepage: http://www.tandfonline.com/loi/ienz20 Dithiocarbamates with potent inhibitory activity against the Saccharomyces cerevisiae β-carbonic anhydrase Murat Bozdag, Fabrizio Carta, Daniela Vullo, Semra Isik, Zeid AlOthman, Sameh M. Osman, Andrea Scozzafava & Claudiu T. Supuran To cite this article: Murat Bozdag, Fabrizio Carta, Daniela Vullo, Semra Isik, Zeid AlOthman, Sameh M. Osman, Andrea Scozzafava & Claudiu T. Supuran (2016) Dithiocarbamates with potent inhibitory activity against the Saccharomyces cerevisiae β-carbonic anhydrase, Journal of Enzyme Inhibition and Medicinal Chemistry, 31:1, 132-136, DOI: 10.3109/14756366.2015.1010529 To link to this article: https://doi.org/10.3109/14756366.2015.1010529 Published online: 11 Feb 2015. Submit your article to this journal Article views: 201 View related articles View Crossmark data Citing articles: 8 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=ienz20 http://informahealthcare.com/enz ISSN: 1475-6366 (print), 1475-6374 (electronic) J Enzyme Inhib Med Chem, 2016; 31(1): 132–136 ! 2015 Informa UK Ltd. DOI: 10.3109/14756366.2015.1010529 RESEARCH ARTICLE Dithiocarbamates with potent inhibitory activity against the Saccharomyces cerevisiae b-carbonic anhydrase Murat Bozdag1, Fabrizio -

Quantitative Metrics for Drug–Target Ligandability, Drug Discov Today (2018)
Drug Discovery Today Volume 00, Number 00 December 2018 REVIEWS POST SCREEN – Quantitative metrics for drug target ligandability Reviews 1 1,2 Sinisa Vukovic and David J. Huggins 1 Department of Physics, Cavendish Laboratory, University of Cambridge, 19 JJ Thomson Avenue, Cambridge CB3 0HE, UK 2 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK Ligandability is a prerequisite for druggability and is a much easier concept to understand, model and predict because it does not depend on the complex pharmacodynamic and pharmacokinetic mechanisms in the human body. In this review, we consider a metric for quantifying ligandability from experimental data. We discuss ligandability in terms of the balance between effort and reward. The metric is evaluated for a standard set of well-studied drug targets – some traditionally considered to be ligandable and some regarded as difficult. We suggest that this metric should be used to systematically improve computational predictions of ligandability, which can then be applied to novel drug targets to predict their tractability. Introduction identified, one of the several computational approaches can be The druggability of a protein target is defined as the relative ease or used to predict the druggability or ligandability of the target. These difficulty of developing a small molecule that can effectively include MAPPOD [13], SiteMap [14], WaterMap [15], fpocket [16], modulate protein activity in vivo [1]. The ligandability of a protein DLID [17], DrugFEATURE [18], DrugPred [19], FTMap [20], JEDI is defined as the relative ease or difficulty of developing a small [21] and others [22,23]. It is worth noting that some methods, such molecule that can bind to the protein in vitro [2]. -

Carbonic Anhydrase II, Human Recombinant, Expressed in Escherichia Coli
Carbonic Anhydrase II, human recombinant, expressed in Escherichia coli Catalog Number C6624 Storage Temperature –20 °C Synonym: CA II Enzyme activity: 1. Conversion of carbon dioxide to bicarbonate: Product Description + Carbonic anhydrases (CA) are a family of enzymes that CO2 + H2O ® HCO3 + H catalyze the rapid conversion of carbon dioxide to bicarbonate and protons, a reaction that occurs rather Specific activity: ³5,000 units/mg slowly in the absence of a catalyst. The active site of most carbonic anhydrases contains a zinc ion. They Unit Definition: One unit will decrease the pH of a 1-2 are, therefore, classified as metalloenzymes. 20 mM Tris buffer from pH 8.3 to 6.3 in 1 minute at 0 °C. Carbonic anhydrases are widely distributed in plant and animal tissues where they are involved in diverse 2. Esterase activity: physiological processes, such as photosynthesis, p-nitrophenyl acetate + H2O ® p-nitrophenol + pH homeostasis, calcification, and bone resorption. acetate There are at least five distinct CA families (a, b, g, d, The increase in the amount of the product, and e). These families have no significant amino acid p-nitrophenol, is measured as absorbance sequence similarity and, in most cases, are thought to increases at 405 nm. be an example of convergent evolution. The a-CAs are found in humans. At least 14 isoforms of a-CA have Specific activity: ³2 nmole/min/mg been identified in vertebrates with different physiological and pathological roles. The enzymes can Precautions and Disclaimer be localized in the cytosol or mitochondria, be This product is for R&D use only, not for drug, membrane bound with extracellular domains, or be household, or other uses. -
![Mode of Substrate Carboxyl Binding to the [4Fe-4S]+ Cluster of Reduced Aconitase As Studied by 170 and 13C Electron-Nuclear Doub](https://docslib.b-cdn.net/cover/9580/mode-of-substrate-carboxyl-binding-to-the-4fe-4s-cluster-of-reduced-aconitase-as-studied-by-170-and-13c-electron-nuclear-doub-3329580.webp)
Mode of Substrate Carboxyl Binding to the [4Fe-4S]+ Cluster of Reduced Aconitase As Studied by 170 and 13C Electron-Nuclear Doub
Proc. Natl. Acad. Sci. USA Vol. 84, pp. 8854-8858, December 1987 Biochemistry Mode of substrate carboxyl binding to the [4Fe-4S]+ cluster of reduced aconitase as studied by 170 and 13C electron-nuclear double resonance spectroscopy (aconitase mechanism/nitro analogues of substrate/isotopic labeling) MARY CLAIRE KENNEDY*, MELANIE WERSTt, JOSHUA TELSERt, MARK H. EMPTAGEt, HELMUT BEINERT*§, AND BRIAN M. HOFFMANt *Department of Biochemistry and 1National Biomedical ESR Center, Medical College of Wisconsin, Milwaukee, WI 53226; tDepartment of Chemistry, Northwestern University, Evanston, IL 60201; and tCentral Research and Development Department, E. I. duPont de Nemours, Wilmington, DE 19898 Contributed by Helmut Beinert, August 25, 1987 ABSTRACT The active form of aconitase has a diamag- ening of the EPR signal in the presence of 170-labeled netic [4Fe-4S]21 cluster. A specific iron ion (Fea, which is lost substrate$ and solvent water indicated that solvent HxO$ during inactivation) is the binding site for substrate, as shown and/or the -OH group of substrate could be bound to the by Mossbauer spectroscopy. We have studied the mode of [4Fe-4S]' cluster (3). The binding of HxO and -OH of an substrate and analogue binding at equilibrium to the paramag- analogue to substrate was demonstrated directly by subse- netic [4Fe-4S]+ cluster of the reduced active form by 170 and quent electron-nuclear double resonance (ENDOR) studies 13C electron-nuclear double resonance spectroscopy with spe- with the reduced enzyme (4). Samples equilibrated with cifically labeled substrates. The data show that with substrate, cis-aconitate in H2170 showed ENDOR signals from HO only the carboxyl at C-2 of the propane backbone is strongly bound to the cluster. -

The Reaction Pathways of Zinc Enzymes and Related Biological Catalysts
2 The Reaction Pathways of Zinc Enzymes and Related Biological Catalysts IVANa BERTINI Department of Chemistry University of Florence CLAUDIO LUCHINAT Institute of Agricultural Chemistry University of Bologna I. INTRODUCTION This chapter deals with metalloenzymes wherein the metal acts mainly as a Lewis acid; i.e., the metal does not change its oxidation state nor, generally, its protein ligands. Changes in the coordination sphere may occur on the side ex posed to solvent. The substrate interacts with protein residues inside the active cavity and/or with the metal ion in order to be activated, so that the reaction can occur. Under these circumstances the catalyzed reactions involve, as central steps with often complex reaction pathways, the following bond-breaking and/or formation pro cesses: peptide hydrolysis H H I I R-C-N-R' + H 0 ~ R-C-O- + H-W-R' (2.1) II 2 II I o 0 H carboxylic ester hydrolysis R-C-O-R' + H 0 ~ R-C-O- + H-O-R' + W (2.2) II 2 II o 0 37 38 2/ THE REACTION PATHWAYS OF ZINC ENZYMES AND RELATED BIOLOGICAL CATALYSTS phosphoric ester hydrolysis 0- 0- I I H-O-P-O-R'+HO ~ H-O-P-O-+H-O-R'+W (2.3) II 2 II o 0 nucleophilic addition of OH- and H- 0 R R H O- II "'- / I I H-O- ---t C ----'" O-C ;O=C <--- H- ~ -O-C-H (2.4) ~ II ----- 0 I I 0 R' R' Scheme (2.3) also pertains to the reactions which need ATP hydrolysis to pro mote endoenergetic reactions.