Structure and Function of Enzymes Involved in Tetrapyrrole Biosynthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Investigation of the Chlorophylls of Selected Prasinophyte Algae
University of Nebraska at Omaha DigitalCommons@UNO Student Work 5-1-1986 An investigation of the chlorophylls of selected prasinophyte algae. Leslie Carlat Kwasnieski Follow this and additional works at: https://digitalcommons.unomaha.edu/studentwork Recommended Citation Kwasnieski, Leslie Carlat, "An investigation of the chlorophylls of selected prasinophyte algae." (1986). Student Work. 3376. https://digitalcommons.unomaha.edu/studentwork/3376 This Thesis is brought to you for free and open access by DigitalCommons@UNO. It has been accepted for inclusion in Student Work by an authorized administrator of DigitalCommons@UNO. For more information, please contact [email protected]. AN INVESTIGATION OF THE CHLOROPHYLLS OF SELECTED PRASINOPHYTE ALGAE A Thesis Presented to the Department of Biology and the Faculty of the Graduate College U n iv e rs ity o f Nebraska In Partial Fulfillment o f the Requirements fo r the Degree Master of Arts University of Nebraska at Omaha by Leslie Carl at Kwasnieski May, 1986 UMI Number: EP74978 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI EP74978 Published by ProQuest LLC (2015). Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code ProQuest ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1345 Ann Arbor, Ml 48105-1346 THESIS ACCEPTANCE Accepted for the faculty of the Graduate College, University of Nebraska, in partial fulfillm ent of the requirements for the degree Master of Arts, University of Nebraska at Omaha. -

Hyperbilirubinemia
Porphyrins Porphyrins (Porphins) are cyclic tetrapyrol compounds formed by the linkage )). of four pyrrole rings through methenyl bridges (( HC In the reduced porphyrins (Porphyrinogens) the linkage of four pyrrole rings (tetrapyrol) through methylene bridges (( CH2 )) The characteristic property of porphyrins is the formation of complexes with the metal ion bound to nitrogen atoms of the pyrrole rings. e.g. Heme (iron porphyrin). Proteins which contain heme ((hemoproteins)) are widely distributed e.g. Hemoglobin, Myoglobin, Cytochromes, Catalase & Tryptophan pyrrolase. Natural porphyrins have substituent side chains on the eight hydrogen atoms numbered on the pyrrole rings. These side chains are: CH 1-Methyl-group (M)… (( 3 )) 2-Acetate-group (A)… (( CH2COOH )) 3-Propionate-group (P)… (( CH2CH2COOH )) 4-Vinyl-group (V)… (( CH CH2 )) Porphyrins with asymmetric arrangement of the side chains are classified as type III porphyrins while those with symmetric arrangement of the side chains are classified as type I porphyrins. Only types I & III are present in nature & type III series is more important because it includes heme. 1 Heme Biosynthesis Heme biosynthesis occurs through the following steps: 1-The starting reaction is the condensation between succinyl-CoA ((derived from citric acid cycle in the mitochondria)) & glycine, this reaction is a rate limiting reaction in the hepatic heme synthesis, it occurs in the mitochondria & is catalyzed by ALA synthase (Aminolevulinate synthase) enzyme in the presence of pyridoxal phosphate as a cofactor. The product of this reaction is α-amino-β-ketoadipate which is rapidly decarboxylated to form δ-aminolevulinate (ALA). 2-In the cytoplasm condensation reaction between two molecules of ALA is catalyzed by ALA dehydratase enzyme to form two molecules of water & one 2 molecule of porphobilinogen (PBG) which is a precursor of pyrrole. -

Levels of 6-Aminolevulinate Dehydratase, Uroporphyrinogen-I Synthase, and Protoporphyrin IX in Erythrocytes from Anemic Mutant M
Proc. Natl. Acad. Sci. USA Vol. 74, No. 3, pp. 1181-1184, March 1977 Genetics Levels of 6-aminolevulinate dehydratase, uroporphyrinogen-I synthase, and protoporphyrin IX in erythrocytes from anemic mutant mice (hemolytic disease/reticulocytosis/heme biosynthesis/development) SHIGERU SASSA* AND SELDON E. BERNSTEINt * The Rockefeller University, New York, N.Y. 10021; and t The Jackson Laboratory, Bar Harbor, Maine 04609 Communicated by Elizabeth S. Russell, December 23, 1976 ABSTRACT Levels of erythrocyte 6-aminolevulinate MATERIALS AND METHODS dehydratase [ALA-dehydratase; porphobilinogen synthase; 5- aminolevulinate hydro-lyase (adding 5-aminolevulinate and Animals. Genetically anemic mutant mice, their littermate cyclizing), EC 4.2.1.241, uroporphyrinogen-I synthase [Uro- controls, and the C57BL/6J and DBA/2J inbred strains were synthase; porphobilinogen ammonia-lyase (polymerizing), EC reared in research colonies of the Jackson Laboratory. The or- 4.3.1.8], and protoporphyrin IX (Proto) were measured by sen- igins and hematological, as well as general, characteristics of sitive semimicroassays using 2-5 IAl of whole blood obtained the various types of anemic mice have been reviewed elsewhere from normal and anemic mutant mice. The levels of erythrocyte (1-3). The mutants employed in these studies of ALA-dehydratase and Uro-synthase showed marked develop- hemolytic mental changes and ALA-dehydratase was influenced by the disease included: jaundice (ja) (4), spherocytosis (sph) (5), he- L v gene. molytic anemia (ha) (1, 6), and normoblastosis (nb) (1, 7). Other Mice with overt hemolytic diseases (ja/ja, sph/sph, nb/nb, anemias used were: microcytosis (mk) (8), Steel (SI) (9), W series ha/ha) had 10- to 20-fold increases in ALA-dehydratase, Uro- (W) (10), and Hertwig's anemia (an) (11). -

Porphyrins & Bile Pigments
Bio. 2. ASPU. Lectu.6. Prof. Dr. F. ALQuobaili Porphyrins & Bile Pigments • Biomedical Importance These topics are closely related, because heme is synthesized from porphyrins and iron, and the products of degradation of heme are the bile pigments and iron. Knowledge of the biochemistry of the porphyrins and of heme is basic to understanding the varied functions of hemoproteins in the body. The porphyrias are a group of diseases caused by abnormalities in the pathway of biosynthesis of the various porphyrins. A much more prevalent clinical condition is jaundice, due to elevation of bilirubin in the plasma, due to overproduction of bilirubin or to failure of its excretion and is seen in numerous diseases ranging from hemolytic anemias to viral hepatitis and to cancer of the pancreas. • Metalloporphyrins & Hemoproteins Are Important in Nature Porphyrins are cyclic compounds formed by the linkage of four pyrrole rings through methyne (==HC—) bridges. A characteristic property of the porphyrins is the formation of complexes with metal ions bound to the nitrogen atom of the pyrrole rings. Examples are the iron porphyrins such as heme of hemoglobin and the magnesium‐containing porphyrin chlorophyll, the photosynthetic pigment of plants. • Natural Porphyrins Have Substituent Side Chains on the Porphin Nucleus The porphyrins found in nature are compounds in which various side chains are substituted for the eight hydrogen atoms numbered in the porphyrin nucleus. As a simple means of showing these substitutions, Fischer proposed a shorthand formula in which the methyne bridges are omitted and a porphyrin with this type of asymmetric substitution is classified as a type III porphyrin. -

UROPORPHYRINOGEN HII COSYNTHETASE in HUMAN Hemolysates from Five Patientswith Congenital Erythropoietic Porphyriawas Much Lower
UROPORPHYRINOGEN HII COSYNTHETASE IN HUMAN CONGENITAL ERYTHROPOIETIC PORPHYRIA * BY GIOVANNI ROMEO AND EPHRAIM Y. LEVIN DEPARTMENT OF PEDIATRICS, THE JOHNS HOPKINS UNIVERSITY SCHOOL OF MEDICINE Communicated by William L. Straus, Jr., April 24, 1969 Abstract.-Activity of the enzyme uroporphyrinogen III cosynthetase in hemolysates from five patients with congenital erythropoietic porphyria was much lower than the activity in control samples. The low cosynthetase activity in patients was not due to the presence of a free inhibitor or some competing en- zymatic activity, because hemolysates from porphyric subjects did not interfere either with the cosynthetase activity of hemolysates from normal subjects or with cosynthetase prepared from hematopoietic mouse spleen. This partial deficiency of cosynthetase in congenital erythropoietic porphyria corresponds to that shown previously in the clinically similar erythropoietic porphyria of cattle and explains the overproduction of uroporphyrin I in the human disease. Erythropoietic porphyria is a rare congenital disorder of man and cattle, characterized by photosensitivity, erythrodontia, hemolytic anemia, and por- phyrinuria.1 Many of the clinical manifestations of the disease can be explained by the production in marrow, deposition in tissues, and excretion in the urine and feces, of large amounts of uroporphyrin I and coproporphyrin I, which are products of the spontaneous oxidation of uroporphyrinogen I and its decarboxyl- ated derivative, coproporphyrinogen I. In cattle, the condition is inherited -

Medications in Pregnant and Nursing Mothers
Medications in Pregnant and Nursing Mothers NADINE M. GIRGIS, OD, FAAO ASSISTANT PROFESSOR YIN C. TEA, OD, FAAO CHIEF, PEDIATRICS AND BINOCULAR VISION ASSISTANT PROFESSOR Gestation age vs fetal age Gestation age-sperm penetrates the egg and zygote is formed Zygote (fertilized egg) travels from fallopian tube to uterus During this time, egg divides into cells - called a morula Continued dividing and morula - called a blastocyst - embeds in the uterus anywhere from 6-12 days after conception This begins the embryonic stage and fetal age begins Fetal development-1st trimester Gestation age week 3-fetal age week 1: a lot of basic growth Brain, spinal cord, heart, GI tract begin development 1st trimester Gestation age-week 4 and 5: embryo ¼ inch long Arm and leg buds, ears, eyes forming Placenta forming and producing hormones Heart is beating at a steady rhythm Movement of rudimentary blood through blood vessels 1st trimester Gestation age week 6: embryo is ½ in length Lungs, jaw, nose, plate formation, hands and feet Hand and feet buds have webbed structures Brain forming into complex parts 1st trimester Gestation age week 7: weighs less than an aspirin All essential organs have begun to form Hair, nail follicles, eyelids and tongue starting to form Trunk begins to straighten out 1st trimester Gestation age week 8: 1 in long, size of a bean All parts of adult are now present in the embryo Bones beginning to form Muscles begin to contract Facial features, including eyelids more developed Gestation age weeks 9-13: 3 in and weighs -

Preferred Drug List
October 2021 Preferred Drug List The Preferred Drug List, administered by CVS Caremark® on behalf of Siemens, is a guide within select therapeutic categories for clients, plan members and health care providers. Generics should be considered the first line of prescribing. If there is no generic available, there may be more than one brand-name medicine to treat a condition. These preferred brand-name medicines are listed to help identify products that are clinically appropriate and cost-effective. Generics listed in therapeutic categories are for representational purposes only. This is not an all-inclusive list. This list represents brand products in CAPS, branded generics in upper- and lowercase Italics, and generic products in lowercase italics. PLAN MEMBER HEALTH CARE PROVIDER Your benefit plan provides you with a prescription benefit program Your patient is covered under a prescription benefit plan administered administered by CVS Caremark. Ask your doctor to consider by CVS Caremark. As a way to help manage health care costs, prescribing, when medically appropriate, a preferred medicine from authorize generic substitution whenever possible. If you believe a this list. Take this list along when you or a covered family member brand-name product is necessary, consider prescribing a brand name sees a doctor. on this list. Please note: Please note: • Your specific prescription benefit plan design may not cover • Generics should be considered the first line of prescribing. certain products or categories, regardless of their appearance in • This drug list represents a summary of prescription coverage. It is this document. Products recently approved by the U.S. Food and not all-inclusive and does not guarantee coverage. -

Specific Pattern of Gene Expression During Induction of Mouse Erythroleukemia Cells
Downloaded from genesdev.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Specific pattern of gene expression during induction of mouse erythroleukemia cells Peter J. Fraser and Peter J. Curtis The Wistar Institute of Anatomy and Biology, Philadelphia, Pennsylvania 19104 USA We have studied the expression of several characterized genes during induction of mouse erythroleukemia (MEL) cells with dimethyl sulfoxide (DMSO) and have observed a specific pattern of changes in transcriptional activity and steady-state RNA levels associated with erythroid differentiation. During induction there is a gradual, steady decrease in total transcriptional activity and RNA content per cell, which by day 3 of DMSO treatment amounts to less than 50% of the level in the uninduced cell. During this time we observe increases in transcriptional activity for 5-aminolevulinic acid synthase, carbonic anhydrase form II, and band 3 coordinate with the large increase in [3-globin gene transcription. The results also demonstrate an early decrease in transcription for carbonic anhydrase form I, which precedes decreases in transcription for glyceraldehyde phosphate dehydrogenase and rRNA genes. Changes in steady-state RNA levels reflected changes in transcriptional activity during induction except for carbonic anhydrase II mRNA. These results represent the first report characterizing the regulated expression at transcriptional and posttranscriptional levels of several known genes that are characteristically expressed in the erythrocyte. The results demonstrate that coordinate gene expression in erythroid differentiation occurs primarily at the level of transcription. [Key Words: Coordinated gene regulation; transcriptional activity; erythroid differentiation] Received June 25, 1987; revised version accepted August 14, 1987. Adult erythroid differentiation requires activation and/ dinately such that they may assemble to form the char- or modulation of many genes whose expression is regu- acteristic erythrocyte cytoskeleton. -
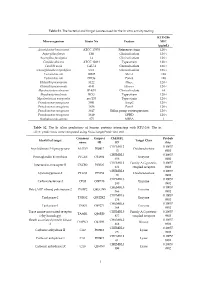
Table S1. the Bacterial and Fungal Isolates Used for the in Vitro Activity Testing
Table S1. The bacterial and fungal isolates used for the in vitro activity testing. KTU-286 Microorganism Strain No Feature MIC (µg/mL) Acinetobacter baummanii ATCC 17978 Reference strain 128 < Aspergillus flavus 12B Clinical isolate 128 < Aspergillus fumigatus 14 Clinical isolate 128 < Candida albicans ATCC 10231 Type strain 128 < Candida auris CAU-1 Clinical isolate 128 < Cuninghamella corymbifera CC1 Clinical isolate 128 < Escherichia coli 10025 Mcr-1 128 Escherichia coli DH5a Pan-S 128 Klebsiella pneumoniae 3122 blaKPC 128 < Klebsiella pneumoniae 4141 blaNDM-1 128 < Mycobacterium abscesus IP-K01 Clinical isolate 64 Mycobacterium bovis BCG Type strain 128 < Mycobacterium smegmatis mc2155 Type strain 128 < Pseudomonas aeruginosa 3691 AmpC 128 < Pseudomonas aeruginosa 3656 Pan-S 128 < Pseudomonas aeruginosa 3647 Efflux pump overexpression 128 < Pseudomonas aeruginosa 3619 OPRD 128 < Staphylococcus aureus 875 MRSA 1 Table S2. The In silico predictions of human proteins interacting with KTU-286. The in silico predictions were computed using SwissTargetPrediction tool. Common Uniprot ChEMBL Probab Identified target Target Class name ID ID ility CHEMBL2 0.10057 Arachidonate 5-lipoxygenase ALOX5 P09917 Oxidoreductase 15 8902 CHEMBL5 0.10057 Prostaglandin E synthase PTGES O14684 Enzyme 658 8902 CHEMBL2 Family A G protein- 0.10057 Interleukin-8 receptor B CXCR2 P25025 434 coupled receptor 8902 CHEMBL2 0.10057 Cyclooxygenase-2 PTGS2 P35354 Oxidoreductase 30 8902 CHEMBL3 0.10057 Carboxylesterase 2 CES2 O00748 Enzyme 180 8902 CHEMBL5 0.10057 Poly -

Structural and Functional Studies of the Light-Dependent Protochlorophyllide Oxidoreductase Enzyme
Structural and Functional Studies of the Light-Dependent Protochlorophyllide Oxidoreductase Enzyme David Robert Armstrong Department of Molecular Biology and Biotechnology A thesis submitted for the degree of Doctor of Philosophy September 2014 Abstract The light-dependent enzyme protochlorophyllide oxidoreductase (POR) is a key enzyme in the chlorophyll biosynthesis pathway, catalysing the reduction of the C17 - C18 bond in protochlorophyllide (Pchlide) to form chlorophyllide (Chlide). This reaction involves the light- induced transfer of a hydride from the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor, followed by proton transfer from a catalytic tyrosine residue. Much work has been done to elucidate the catalytic mechanism of POR, however little is known about the protein structure. POR isoforms in plants are also notable as components of the prolamellar bodies (PLBs), large paracrystalline structures that are precursors to the thylakoid membranes in mature chloroplasts. Bioinformatics studies have identified a number of proteins, related to POR, which contain similar structural features, leading to the production of a structural model for POR. A unique loop region of POR was shown by EPR to be mobile, with point mutations within this region causing a reduction in enzymatic activity. Production of a 2H, 13C, 15N-labelled sample of POR for NMR studies has enabled significant advancement in the understanding of the protein structure. This includes the calculation of backbone torsion angles for the majority of the protein, in addition to the identification of multiple dynamic regions of the protein. The protocol for purification of Pchlide, the substrate for POR, has been significantly improved, providing high quality pigment for study of the POR ternary complex. -

Targeting Mycobacterium Tuberculosis Proteins: Structure and Function Studies of Five Essential Proteins
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 411 Targeting Mycobacterium tuberculosis Proteins: Structure and Function Studies of Five Essential Proteins ADRIAN SUAREZ COVARRUBIAS ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 UPPSALA ISBN 978-91-554-7134-7 2008 urn:nbn:se:uu:diva-8580 !! ""# !$"" % & % % '& &( )& * + &( ,- ( ""#( ) ' $ , , % + ' ( ( !!( .! ( ( /,0 12#31!34432!32( )& & & % - & &- % % 56 ( )& & % & % & % %% & ( )& % & 5 6 7!# 74## & (" !(2 8 ( 74## 7!# 93 & & * * %% % 3 - ( 7!14 & & *& && : & & (4 8 *&& % & & & ( & &; & %% ( < % & % & 5 6( )& & % - & * & % ( 7".. % ; * 3% & 93& 3 ' & *&& % &( 3 *& ( 44= % & ; * % ( 722# % & &3 & !(# 8 ( , & % & % & * ( &* ( & & & , // 7".. 7!# 7!14 74## 722# >3 & ! " # " #$ %&'" " ()*%+,- " ? ,- ""# /,,0 !.4!3.! /,0 12#31!34432!32 $ $$$ 3#4#" 5& $@@ (;(@ A B $ $$$ 3#4#"6 List of publications This thesis consists of a comprehensive summary based on the following papers. In the text, the papers will be referred to by their roman numerals. I Covarrubias, A. S., Larsson, A. M., Högbom, M., Lindberg, J., Bergfors, T., Björkelid, C., Mowbray, S. L., Unge, T., and Jones, T. A. (2005). Structure and -

Significance of Heme and Heme Degradation in the Pathogenesis Of
International Journal of Molecular Sciences Review Significance of Heme and Heme Degradation in the Pathogenesis of Acute Lung and Inflammatory Disorders Stefan W. Ryter Proterris, Inc., Boston, MA 02118, USA; [email protected] Abstract: The heme molecule serves as an essential prosthetic group for oxygen transport and storage proteins, as well for cellular metabolic enzyme activities, including those involved in mitochondrial respiration, xenobiotic metabolism, and antioxidant responses. Dysfunction in both heme synthesis and degradation pathways can promote human disease. Heme is a pro-oxidant via iron catalysis that can induce cytotoxicity and injury to the vascular endothelium. Additionally, heme can modulate inflammatory and immune system functions. Thus, the synthesis, utilization and turnover of heme are by necessity tightly regulated. The microsomal heme oxygenase (HO) system degrades heme to carbon monoxide (CO), iron, and biliverdin-IXα, that latter which is converted to bilirubin-IXα by biliverdin reductase. Heme degradation by heme oxygenase-1 (HO-1) is linked to cytoprotection via heme removal, as well as by activity-dependent end-product generation (i.e., bile pigments and CO), and other potential mechanisms. Therapeutic strategies targeting the heme/HO-1 pathway, including therapeutic modulation of heme levels, elevation (or inhibition) of HO-1 protein and activity, and application of CO donor compounds or gas show potential in inflammatory conditions including sepsis and pulmonary diseases. Keywords: acute lung injury; carbon monoxide; heme; heme oxygenase; inflammation; lung dis- ease; sepsis Citation: Ryter, S.W. Significance of Heme and Heme Degradation in the Pathogenesis of Acute Lung and Inflammatory Disorders. Int. J. Mol. 1. Introduction Sci.