CHAPTER 12 Molecular Spectroscopy 1: Rotational and Vibrational Spectra
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions
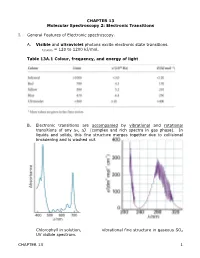
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. εphoton = 120 to 1200 kJ/mol. Table 13A.1 Colour, frequency, and energy of light B. Electronic transitions are accompanied by vibrational and rotational transitions of any Δν, ΔJ (complex and rich spectra in gas phase). In liquids and solids, this fine structure merges together due to collisional broadening and is washed out. Chlorophyll in solution, vibrational fine structure in gaseous SO2 UV visible spectrum. CHAPTER 13 1 C. Molecules need not have permanent dipole to undergo electronic transitions. All that is required is a redistribution of electronic and nuclear charge between the initial and final electronic state. Transition intensity ~ |µ|2 where µ is the magnitude of the transition dipole moment given by: ˆ d µ = ∫ ψfinalµψ initial τ where µˆ = −e ∑ri + e ∑Ziri € electrons nuclei r vector position of the particle i = D. Franck-Condon principle: Because nuclei are so much more massive and € sluggish than electrons, electronic transitions can happen much faster than the nuclei can respond. Electronic transitions occur vertically on energy diagram at right. (Hence the name vertical transition) Transition probability depends on vibrational wave function overlap (Franck-Condon factor). In this example: ν = 0 ν’ = 0 have small overlap ν = 0 ν’ = 2 have greatest overlap Now because the total molecular wavefunction can be approximately factored into 2 terms, electronic wavefunction and the nuclear position wavefunction (using Born-Oppenheimer approx.) the transition dipole can also be factored into 2 terms also: S µ = µelectronic i,f S d Franck Condon factor i,j i,f = ∫ ψf,vibr ψi,vibr τ = − CHAPTER 13 2 See derivation “Justification 14.2” II. -

Magnetism, Angular Momentum, and Spin
Chapter 19 Magnetism, Angular Momentum, and Spin P. J. Grandinetti Chem. 4300 P. J. Grandinetti Chapter 19: Magnetism, Angular Momentum, and Spin In 1820 Hans Christian Ørsted discovered that electric current produces a magnetic field that deflects compass needle from magnetic north, establishing first direct connection between fields of electricity and magnetism. P. J. Grandinetti Chapter 19: Magnetism, Angular Momentum, and Spin Biot-Savart Law Jean-Baptiste Biot and Félix Savart worked out that magnetic field, B⃗, produced at distance r away from section of wire of length dl carrying steady current I is 휇 I d⃗l × ⃗r dB⃗ = 0 Biot-Savart law 4휋 r3 Direction of magnetic field vector is given by “right-hand” rule: if you point thumb of your right hand along direction of current then your fingers will curl in direction of magnetic field. current P. J. Grandinetti Chapter 19: Magnetism, Angular Momentum, and Spin Microscopic Origins of Magnetism Shortly after Biot and Savart, Ampére suggested that magnetism in matter arises from a multitude of ring currents circulating at atomic and molecular scale. André-Marie Ampére 1775 - 1836 P. J. Grandinetti Chapter 19: Magnetism, Angular Momentum, and Spin Magnetic dipole moment from current loop Current flowing in flat loop of wire with area A will generate magnetic field magnetic which, at distance much larger than radius, r, appears identical to field dipole produced by point magnetic dipole with strength of radius 휇 = | ⃗휇| = I ⋅ A current Example What is magnetic dipole moment induced by e* in circular orbit of radius r with linear velocity v? * 휋 Solution: For e with linear velocity of v the time for one orbit is torbit = 2 r_v. -

Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO
Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO. WHEN we solve the Schrödinger equation, we always obtain two things: 1. a set of eigenstates, |ψn>. 2. a set of eigenstate energies, En QM predicts the existence of discrete, evenly spaced, vibrational energy levels for the SHO. n = 0,1,2,3... For the ground state (n=0), E = ½hν. Notes: This is called the zero point energy. Optical selection rule -- SHO can absorb or emit light with a ∆n = ±1 The IR absorption spectrum for a diatomic molecule, such as HCl: diatomic Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion. (Note: μ here means dipole moment). If a more realistic Morse potential is used in the Schrödinger Equation, these energy levels get scrunched together... ΔE=hν ΔE<hν The vibrational spectroscopy of polyatomic molecules gets more interesting... For diatomic or linear molecules: 3N-5 modes For nonlinear molecules: 3N-6 modes N = number of atoms in molecule The vibrational spectroscopy of polyatomic molecules gets more interesting... Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 All bands are observed in the IR spectrum. -

Lecture 17 Highlights
Lecture 23 Highlights The phenomenon of Rabi oscillations is very different from our everyday experience of how macroscopic objects (i.e. those made up of many atoms) absorb electromagnetic radiation. When illuminated with light (like French fries at McDonalds) objects tend to steadily absorb the light and heat up to a steady state temperature, showing no signs of oscillation with time. We did a calculation of the transition probability of an atom illuminated with a broad spectrum of light and found that the probability increased linearly with time, leading to a constant transition rate. This difference comes about because the many absorption processes at different frequencies produce a ‘smeared-out’ response that destroys the coherent Rabi oscillations and results in a classical incoherent absorption. Selection Rules All of the transition probabilities are proportional to “dipole matrix elements,” such as x jn above. These are similar to dipole moment calculations for a charge distribution in classical physics, except that they involve the charge distribution in two different states, connected by the dipole operator for the transition. In many cases these integrals are zero because of symmetries of the associated wavefunctions. This gives rise to selection rules for possible transitions under the dipole approximation (atom size much smaller than the electromagnetic wave wavelength). For example, consider the matrix element z between states of the n,l , m ; n ', l ', m ' Hydrogen atom labeled by the quantum numbers n,,l m and n',','l m : z = ψ()()r z ψ r d3 r n,l , m ; n ', l ', m ' ∫∫∫ n,,l m n',l ', m ' To make progress, first consider just the φ integral. -

Introduction to Differential Equations
Quantum Dynamics – Quick View Concepts of primary interest: The Time-Dependent Schrödinger Equation Probability Density and Mixed States Selection Rules Transition Rates: The Golden Rule Sample Problem Discussions: Tools of the Trade Appendix: Classical E-M Radiation POSSIBLE ADDITIONS: After qualitative section, do the two state system, and then first and second order transitions (follow Fitzpatrick). Chain together the dipole rules to get l = 2,0,-2 and m = -2, -2, … , 2. ??Where do we get magnetic rules? Look at the canonical momentum and the Asquared term. Schrödinger, Erwin (1887-1961) Austrian physicist who invented wave mechanics in 1926. Wave mechanics was a formulation of quantum mechanics independent of Heisenberg's matrix mechanics. Like matrix mechanics, wave mechanics mathematically described the behavior of electrons and atoms. The central equation of wave mechanics is now known as the Schrödinger equation. Solutions to the equation provide probability densities and energy levels of systems. The time-dependent form of the equation describes the dynamics of quantum systems. http://scienceworld.wolfram.com/biography/Schroedinger.html © 1996-2006 Eric W. Weisstein www-history.mcs.st-andrews.ac.uk/Biographies/Schrodinger.html Quantum Dynamics: A Qualitative Introduction Introductory quantum mechanics focuses on time-independent problems leaving Contact: [email protected] dynamics to be discussed in the second term. Energy eigenstates are characterized by probability density distributions that are time-independent (static). There are examples of time-dependent behavior that are by demonstrated by rather simple introductory problems. In the case of a particle in an infinite well with the range [ 0 < x < a], the mixed state below exhibits time-dependence. -

Oscillator Strength ( F ): Quantum Mechanical Model
Oscillator strength ( f ): quantum mechanical model • For an electronic transition to occur an oscillating dipole must be induced by interaction of the molecules electric field with electromagnetic radiation. 0 • In fact both ε and k can be related to the transition dipole moment (µµµge ) • If two equal and opposite electrical charges (e) are separated by a vectorial distance (r), a dipole moment (µµµ ) of magnitude equal to er is created. µµµ = e r (e = electron charge, r = extent of charge displacement) • The magnitude of charge separation, as the electron density is redistributed in an electronically excited state, is determined by the polarizability (αααα) of the electron cloud which is defined by the transition dipole moment (µµµge ) α = µµµge / E (E = electrical force) µµµge = e r • The magnitude of the oscillator strength ( f ) for an electronic transition is proportional to the square of the transition dipole moment produced by the action of electromagnetic radiation on an electric dipole. 2 2 f ∝ µµµge = ( e r) fobs = fmax ( fe fv fs ) 2 f ∝ µµµge fobs = observed oscillator strength f ∝ ΛΏΦ ∆̅ !2#( fmax = ideal oscillator strength ( ∼1) f = orbital configuration factor ͯͥ e f ∝ ͤ͟ ̅ ͦ Γ fv = vibrational configuration factor fs = spin configuration factor • There are two major contributions to the electronic factor fe : Poor overlap: weak mixing of electronic wavefunctions, e.g. <nπ*>, due to poor spatial overlap of orbitals involved in the electronic transition, e.g. HOMO→LUMO. Symmetry forbidden: even if significant spatial overlap of orbitals exists, the resonant photon needs to induce a large transition dipole moment. -

Spontaneous Rotational Symmetry Breaking in a Kramers Two- Level System
Spontaneous Rotational Symmetry Breaking in a Kramers Two- Level System Mário G. Silveirinha* (1) University of Lisbon–Instituto Superior Técnico and Instituto de Telecomunicações, Avenida Rovisco Pais, 1, 1049-001 Lisboa, Portugal, [email protected] Abstract Here, I develop a model for a two-level system that respects the time-reversal symmetry of the atom Hamiltonian and the Kramers theorem. The two-level system is formed by two Kramers pairs of excited and ground states. It is shown that due to the spin-orbit interaction it is in general impossible to find a basis of atomic states for which the crossed transition dipole moment vanishes. The parametric electric polarizability of the Kramers two-level system for a definite ground-state is generically nonreciprocal. I apply the developed formalism to study Casimir-Polder forces and torques when the two-level system is placed nearby either a reciprocal or a nonreciprocal substrate. In particular, I investigate the stable equilibrium orientation of the two-level system when both the atom and the reciprocal substrate have symmetry of revolution about some axis. Surprisingly, it is found that when chiral-type dipole transitions are dominant the stable ground state is not the one in which the symmetry axes of the atom and substrate are aligned. The reason is that the rotational symmetry may be spontaneously broken by the quantum vacuum fluctuations, so that the ground state has less symmetry than the system itself. * To whom correspondence should be addressed: E-mail: [email protected] -1- I. Introduction At the microscopic level, physical systems are generically ruled by time-reversal invariant Hamiltonians [1]. -

PHYSICAL SPECTROSCOPY) MODULE No. : 5 (TRANSITION PROBABILITIES and TRANSITION DIPOLE MOMENT. OVERVIEW of SELECTION RULES
____________________________________________________________________________________________________ Subject Chemistry Paper No and Title 8 and Physical Spectroscopy Module No and Title 5 and Transition probabilities and transition dipole moment, Overview of selection rules Module Tag CHE_P8_M5 CHEMISTRY PAPER No. : 8 (PHYSICAL SPECTROSCOPY) MODULE No. : 5 (TRANSITION PROBABILITIES AND TRANSITION DIPOLE MOMENT. OVERVIEW OF SELECTION RULES) ____________________________________________________________________________________________________ TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Transition Moment Integral 4. Overview of Selection Rules 5. Summary CHEMISTRY PAPER No. : 8 (PHYSICAL SPECTROSCOPY) MODULE No. : 5 (TRANSITION PROBABILITIES AND TRANSITION DIPOLE MOMENT. OVERVIEW OF SELECTION RULES) ____________________________________________________________________________________________________ 1. Learning Outcomes After studying this module, • you shall be able to understand the basis of selection rules in spectroscopy • Get an idea about how they are deduced. 2. Introduction The intensity of a transition is proportional to the difference in the populations of the initial and final levels, the transition probabilities given by Einstein’s coefficients of induced absorption and emission and to the energy density of the incident radiation. We now examine the Einstein’s coefficients in some detail. 3. Transition Dipole Moment Detailed algebra involving the time-dependent perturbation theory allows us to derive a theoretical expression for the Einstein coefficient of induced absorption ! 2 3 M ij 8π ! 2 B M ij = 2 = 2 ij 6ε 0" 3h (4πε 0 ) where the radiation density is expressed in units of Hz. ! The quantity M ij is known as the transition moment integral, having the same unit as dipole moment, i.e. C m. Apparently, if this quantity is zero for a particular transition, the transition probability will be zero, or, in other words, the transition is forbidden. -

HJ Lipkin the Weizmann Institute of Science Rehovot, Israel
WHO UNDERSTANDS THE ZWEIG-IIZUKA RULE? H.J. Lipkin The Weizmann Institute of Science Rehovot, Israel Abstract: The theoretical basis of the Zweig-Iizuka Ru le is discussed with the emphasis on puzzles and paradoxes which remain open problems . Detailed analysis of ZI-violating transitions arising from successive ZI-allowed transitions shows that ideal mixing and additional degeneracies in the particle spectrum are required to enable cancellation of these higher order violations. A quantitative estimate of the observed ZI-violating f' + 2rr decay be second order transitions via the intermediate KK state agrees with experiment when only on-shell contributions are included . This suggests that ZI-violations for old particles are determined completely by on-shell contributions and cannot be used as input to estimate ZI-violation for new particle decays where no on-shell intermediate states exist . 327 INTRODUCTION - SOME BASIC QUESTIONS 1 The Zweig-Iizuka rule has entered the folklore of particle physics without any clear theoretical understanding or justification. At the present time nobody really understands it, and anyone who claims to should not be believed. Investigating the ZI rule for the old particles raises many interesting questions which may lead to a better understanding of strong interactions as well as giving additional insight into the experimentally observed suppression of new particle decays attributed to the ZI rule. This talk follows an iconoclastic approach emphasizing embarrassing questions with no simple answers which might lead to fruitful lines of investigation. Both theoretical and experimental questions were presented in the talk. However, the experimental side is covered in a recent paper2 and is not duplicated here. -

Chapter 14 Radiating Dipoles in Quantum Mechanics
Chapter 14 Radiating Dipoles in Quantum Mechanics P. J. Grandinetti Chem. 4300 P. J. Grandinetti Chapter 14: Radiating Dipoles in Quantum Mechanics P. J. Grandinetti Chapter 14: Radiating Dipoles in Quantum Mechanics Electric dipole moment vector operator Electric dipole moment vector operator for collection of charges is ∑N ⃗̂휇 ⃗̂ = qkr k=1 Single charged quantum particle bound in some potential well, e.g., a negatively charged electron bound to a positively charged nucleus, would be [ ] ⃗̂휇 ⃗̂ ̂⃗ ̂⃗ ̂⃗ = *qer = *qe xex + yey + zez Expectation value for electric dipole moment vector in Ψ(⃗r; t/ state is ( ) ê ⃗휇 ë < ⃗; ⃗̂휇 ⃗; 휏 < ⃗; ⃗̂ ⃗; 휏 .t/ = Ê Ψ .r t/ Ψ(r t/d = Ê Ψ .r t/ *qer Ψ(r t/d V V Here, d휏 = dx dy dz P. J. Grandinetti Chapter 14: Radiating Dipoles in Quantum Mechanics Time dependence of electric dipole moment Energy Eigenstate Starting with ( ) ê ⃗휇 ë < ⃗; ⃗̂ ⃗; 휏 .t/ = Ê Ψ .r t/ *qer Ψ(r t/d V For a system in eigenstate of Hamiltonian, where wave function has the form, ` ⃗; ⃗ *iEnt_ Ψn.r t/ = n.r/e Electric dipole moment expectation value is ( ) ` ` ê ⃗휇 ë < ⃗ iEnt_ ⃗̂ ⃗ *iEnt_ 휏 .t/ = Ê n .r/e *qer n.r/e d V Time dependent exponential terms cancel out leaving us with ( ) ê ⃗휇 ë < ⃗ ⃗̂ ⃗ 휏 .t/ = Ê n .r/ *qer n.r/d No time dependence!! V No bound charged quantum particle in energy eigenstate can radiate away energy as light or at least it appears that way – Good news for Rutherford’s atomic model. -

Decoupling Light and Matter: Permanent Dipole Moment Induced Collapse of Rabi Oscillations
Decoupling light and matter: permanent dipole moment induced collapse of Rabi oscillations Denis G. Baranov,1, 2, ∗ Mihail I. Petrov,3 and Alexander E. Krasnok3, 4 1Department of Physics, Chalmers University of Technology, 412 96 Gothenburg, Sweden 2Moscow Institute of Physics and Technology, 9 Institutskiy per., Dolgoprudny 141700, Russia 3ITMO University, St. Petersburg 197101, Russia 4Department of Electrical and Computer Engineering, The University of Texas at Austin, Austin, Texas 78712, USA (Dated: June 29, 2018) Rabi oscillations is a key phenomenon among the variety of quantum optical effects that manifests itself in the periodic oscillations of a two-level system between the ground and excited states when interacting with electromagnetic field. Commonly, the rate of these oscillations scales proportionally with the magnitude of the electric field probed by the two-level system. Here, we investigate the interaction of light with a two-level quantum emitter possessing permanent dipole moments. The semi-classical approach to this problem predicts slowing down and even full suppression of Rabi oscillations due to asymmetry in diagonal components of the dipole moment operator of the two- level system. We consider behavior of the system in the fully quantized picture and establish the analytical condition of Rabi oscillations collapse. These results for the first time emphasize the behavior of two-level systems with permanent dipole moments in the few photon regime, and suggest observation of novel quantum optical effects. I. INTRODUCTION ment modifies multi-photon absorption rates. Emission spectrum features of quantum systems possessing perma- Theory of a two-level system (TLS) interacting with nent dipole moments were studied in Refs. -

UV-Vis. Molecular Absorption Spectroscopy
UV-Vis. Molecular Absorption Spectroscopy Prof. Tarek A. Fayed UV-Vis. Electronic Spectroscopy The interaction of molecules with ultraviolet and visible light may results in absorption of photons. This results in electronic transition, involving valance electrons, from ground state to higher electronic states (called excited states). The promoted electrons are electrons of the highest molecular orbitals HOMO. Absorption of ultraviolet and visible radiation in organic molecules is restricted to certain functional groups (known as chromophores) that contain valence electrons of low excitation energy. A chromophore is a chemical entity embedded within a molecule that absorbs radiation at the same wavelength in different molecules. Examples of Chromophores are dienes, aromatics, polyenes and conjugated ketones, etc. Types of electronic transitions Electronic transitions that can take place are of three types which can be considered as; Transitions involving p-, s-, and n-electrons. Transitions involving charge-transfer electrons. Transitions involving d- and f-electrons in metal complexes. Most absorption spectroscopy of organic molecules is based on transitions of n- or -electrons to the *-excited state. These transitions fall in an experimentally convenient region of the spectrum (200 - 700 nm), and need an unsaturated group in the molecule to provide the -electrons. In vacuum UV or far UV (λ<190 nm ) In UV/VIS For formaldehyde molecule; Selection Rules of electronic transitions Electronic transitions may be allowed or forbidden transitions, as reflected by appearance of an intense or weak band according to the magnitude of εmax, and is governed by the following selection rules : 1. Spin selection rule (△S = 0 for the transition to be allowed): there should be no change in spin orientation i.