Transcriptomic Analysis of Field-Droughted Sorghum From
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

Nucleotide Sequence Surrounding a Ribonuclease III Processing Site In
Proc. Nati. Acad. Sci. USA Vol. 74, No. 3, pp. 984-988, March 1977 Biochemistry Nucleotide sequence surrounding a ribonuclease III processing site in bacteriophage T7 RNA (intercistronic region/polycistronic mRNA precursor/hairpin structure/endoribonuclease III) MARTIN ROSENBERG* AND RICHARD A. KRAMERt f * Laboratory of Molecular Biology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20014; and t Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305 Communicated by Charles Yanofsky, January 3, 1977 ABSTRACT We have determined a nucleotide sequence of the 5' end of the gene 0.7 mRNAs, (ii) some fragments that 87 residues surrounding a ribonuclease III (endoribonuclease contain the RNase III cleavage site are not recognized and III; EC 3.1.4.24) processing site in the bacteriophage 17 inter- cistronic region between early genes 0.3 and 0.7. The structural cleaved by the enzyme; and (iii) the 3'-terminal oligoadenylate requirements necessary for proper recognition and cleavage by sequences found on the ends of the in vdvo T7 early mRNAs (11) RNase III are discussed. In addition, other structural features are not encoded by the genome, but presumably represent a characteristic of this intercistronic boundary are described. nontemplate-dependent post-transcriptional modification. Here, we report the complete nucleotide sequence of the gene When bacteriophage T7 infects Escherichia coli, the host RNA 0.3-0.7 intercistronic region of T7 and propose a specific role polymerase (RNA nucleotidyltransferase, EC 2.7.7.6) tran- for RNA secondary structure in substrate recognition and action scribes only the early region of the phage genome (i.e., leftmost of RNase III. -

Insights Into Anaerobic Degradation of Benzene and Naphthalene
TECHNISCHE UNIVERSITÄT MÜNCHEN Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt Lehrstuhl für Siedlungswasserwirtschaft Insights into anaerobic degradation of benzene and naphthalene Xiyang Dong Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzende(r): Prof. Dr. Wolfgang Liebl Prüfer der Dissertation: 1. Priv.-Doz. Dr. Tillmann Lueders 2. Prof. Dr. Siegfried Scherer 3. Prof. Dr. Rainer Meckenstock Die Dissertation wurde am 03.07.2017 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 18.08.2017 angenommen. 天道酬勤 God rewards those who work hard Abstract Aromatic hydrocarbons, e.g. benzene and naphthalene, have toxic, mutagenic and/or carcinogenic properties. Fortunately, these compounds can be degraded in environmental systems by indigenous microorganisms. Especially under anaerobic conditions, the physiology and ecology of the microbes involved are still poorly understood. In this thesis, important knowledge gaps are addressed in this field using microbiological approaches combined with “omics tools” (metagenomics and metaproteomics). A novel “reverse stable isotope labelling” approach is also introduced to investigate biodegradation activities. Firstly, the enrichment culture BPL was studied, which can degrade benzene coupled with sulfate reduction. It is dominated by an organism of the genus Pelotomaculum. Members of this genus are usually known to be fermenters, undergoing syntrophy with anaerobic respiring microorganisms or methanogens. It remains unclear if Pelotomaculum identified here (namely, Pelotomaculum candidate BPL) could perform both benzene degradation and sulfate reduction. By using a metagenomic approach, a high-quality genome was reconstructed for it. -

Arxiv:Q-Bio.QM/0511042 V2 25 Nov 2005 Nomto Ae Lseig Upeetr Material Supplementary Clustering: Based Information .THE I
Information based clustering: Supplementary material Noam Slonim, Gurinder Singh Atwal, Gaˇsper Tkaˇcik, and William Bialek Joseph Henry Laboratories of Physics, and Lewis–Sigler Institute for Integrative Genomics Princeton University, Princeton, New Jersey 08544 USA (Dated: December 4, 2005) This technical report provides the supplementary material for a paper entitled “Information based clustering,” to appear shortly in Proceedings of the National Academy of Sciences (USA). In Section I we present in detail the iterative clustering algorithm used in our experiments and in Section II we describe the validation scheme used to determine the statistical significance of our results. Then in subsequent sections we provide all the experimental results for three very different applications: the response of gene expression in yeast to different forms of environmental stress, the dynamics of stock prices in the Standard and Poor’s 500, and viewer ratings of popular movies. In particular, we highlight some of the results that seem to deserve special attention. All the experimental results and relevant code, including a freely available web application, can be found at http://www.genomics.princeton.edu/biophysics-theory . Contents many choices at different levels of the analysis. In recent work we suggest that some generality can be achieved I. The Iclust algorithm 1 through the use of information theory (1). Here we re- view this formulation briefly and then proceed to the II. Evaluating clusters’ coherence 3 technical details of its implementation that were left out III. First application: The yeast ESR data 3 of Ref (1). A. Description of the data 3 We formulate clustering as a tradeoff between maxi- B. -
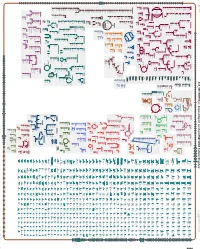
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -
Figure S1. Reverse Transcription‑Quantitative PCR Analysis of ETV5 Mrna Expression Levels in Parental and ETV5 Stable Transfectants
Figure S1. Reverse transcription‑quantitative PCR analysis of ETV5 mRNA expression levels in parental and ETV5 stable transfectants. (A) Hec1a and Hec1a‑ETV5 EC cell lines; (B) Ishikawa and Ishikawa‑ETV5 EC cell lines. **P<0.005, unpaired Student's t‑test. EC, endometrial cancer; ETV5, ETS variant transcription factor 5. Figure S2. Survival analysis of sample clusters 1‑4. Kaplan Meier graphs for (A) recurrence‑free and (B) overall survival. Survival curves were constructed using the Kaplan‑Meier method, and differences between sample cluster curves were analyzed by log‑rank test. Figure S3. ROC analysis of hub genes. For each gene, ROC curve (left) and mRNA expression levels (right) in control (n=35) and tumor (n=545) samples from The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer cohort are shown. mRNA levels are expressed as Log2(x+1), where ‘x’ is the RSEM normalized expression value. ROC, receiver operating characteristic. Table SI. Clinicopathological characteristics of the GSE17025 dataset. Characteristic n % Atrophic endometrium 12 (postmenopausal) (Control group) Tumor stage I 91 100 Histology Endometrioid adenocarcinoma 79 86.81 Papillary serous 12 13.19 Histological grade Grade 1 30 32.97 Grade 2 36 39.56 Grade 3 25 27.47 Myometrial invasiona Superficial (<50%) 67 74.44 Deep (>50%) 23 25.56 aMyometrial invasion information was available for 90 of 91 tumor samples. Table SII. Clinicopathological characteristics of The Cancer Genome Atlas Uterine Corpus Endometrioid Cancer dataset. Characteristic n % Solid tissue normal 16 Tumor samples Stagea I 226 68.278 II 19 5.740 III 70 21.148 IV 16 4.834 Histology Endometrioid 271 81.381 Mixed 10 3.003 Serous 52 15.616 Histological grade Grade 1 78 23.423 Grade 2 91 27.327 Grade 3 164 49.249 Molecular subtypeb POLE 17 7.328 MSI 65 28.017 CN Low 90 38.793 CN High 60 25.862 CN, copy number; MSI, microsatellite instability; POLE, DNA polymerase ε. -

The Metabolic Building Blocks of a Minimal Cell Supplementary
The metabolic building blocks of a minimal cell Mariana Reyes-Prieto, Rosario Gil, Mercè Llabrés, Pere Palmer and Andrés Moya Supplementary material. Table S1. List of enzymes and reactions modified from Gabaldon et. al. (2007). n.i.: non identified. E.C. Name Reaction Gil et. al. 2004 Glass et. al. 2006 number 2.7.1.69 phosphotransferase system glc + pep → g6p + pyr PTS MG041, 069, 429 5.3.1.9 glucose-6-phosphate isomerase g6p ↔ f6p PGI MG111 2.7.1.11 6-phosphofructokinase f6p + atp → fbp + adp PFK MG215 4.1.2.13 fructose-1,6-bisphosphate aldolase fbp ↔ gdp + dhp FBA MG023 5.3.1.1 triose-phosphate isomerase gdp ↔ dhp TPI MG431 glyceraldehyde-3-phosphate gdp + nad + p ↔ bpg + 1.2.1.12 GAP MG301 dehydrogenase nadh 2.7.2.3 phosphoglycerate kinase bpg + adp ↔ 3pg + atp PGK MG300 5.4.2.1 phosphoglycerate mutase 3pg ↔ 2pg GPM MG430 4.2.1.11 enolase 2pg ↔ pep ENO MG407 2.7.1.40 pyruvate kinase pep + adp → pyr + atp PYK MG216 1.1.1.27 lactate dehydrogenase pyr + nadh ↔ lac + nad LDH MG460 1.1.1.94 sn-glycerol-3-phosphate dehydrogenase dhp + nadh → g3p + nad GPS n.i. 2.3.1.15 sn-glycerol-3-phosphate acyltransferase g3p + pal → mag PLSb n.i. 2.3.1.51 1-acyl-sn-glycerol-3-phosphate mag + pal → dag PLSc MG212 acyltransferase 2.7.7.41 phosphatidate cytidyltransferase dag + ctp → cdp-dag + pp CDS MG437 cdp-dag + ser → pser + 2.7.8.8 phosphatidylserine synthase PSS n.i. cmp 4.1.1.65 phosphatidylserine decarboxylase pser → peta PSD n.i. -

Monilinia Fructicola, Monilinia Laxa and Monilinia Fructigena, the Causal Agents of Brown Rot on Stone Fruits Rita M
De Miccolis Angelini et al. BMC Genomics (2018) 19:436 https://doi.org/10.1186/s12864-018-4817-4 RESEARCH ARTICLE Open Access De novo assembly and comparative transcriptome analysis of Monilinia fructicola, Monilinia laxa and Monilinia fructigena, the causal agents of brown rot on stone fruits Rita M. De Miccolis Angelini* , Domenico Abate, Caterina Rotolo, Donato Gerin, Stefania Pollastro and Francesco Faretra Abstract Background: Brown rots are important fungal diseases of stone and pome fruits. They are caused by several Monilinia species but M. fructicola, M. laxa and M. fructigena are the most common all over the world. Although they have been intensively studied, the availability of genomic and transcriptomic data in public databases is still scant. We sequenced, assembled and annotated the transcriptomes of the three pathogens using mRNA from germinating conidia and actively growing mycelia of two isolates of opposite mating types per each species for comparative transcriptome analyses. Results: Illumina sequencing was used to generate about 70 million of paired-end reads per species, that were de novo assembled in 33,861 contigs for M. fructicola, 31,103 for M. laxa and 28,890 for M. fructigena. Approximately, 50% of the assembled contigs had significant hits when blasted against the NCBI non-redundant protein database and top-hits results were represented by Botrytis cinerea, Sclerotinia sclerotiorum and Sclerotinia borealis proteins. More than 90% of the obtained sequences were complete, the percentage of duplications was always less than 14% and fragmented and missing transcripts less than 5%. Orthologous transcripts were identified by tBLASTn analysis using the B. -

Unknown Areas of Activity of Human Ribonuclease Dicer: a Putative Deoxyribonuclease Activity
molecules Article Unknown Areas of Activity of Human Ribonuclease Dicer: A Putative Deoxyribonuclease Activity Marta Wojnicka , Agnieszka Szczepanska and Anna Kurzynska-Kokorniak * Department of Ribonucleoprotein Biochemistry, Institute of Bioorganic Chemistry Polish Academy of Sciences, 61-704 Poznan, Poland; [email protected] (M.W.); [email protected] (A.S.) * Correspondence: [email protected] Received: 31 January 2020; Accepted: 17 March 2020; Published: 20 March 2020 Abstract: The Dicer ribonuclease plays a crucial role in the biogenesis of small regulatory RNAs (srRNAs) by processing long double-stranded RNAs and single-stranded hairpin RNA precursors into small interfering RNAs (siRNAs) and microRNAs (miRNAs), respectively. Dicer-generated srRNAs can control gene expression by targeting complementary transcripts and repressing their translation or inducing their cleavage. Human Dicer (hDicer) is a multidomain enzyme comprising a putative helicase domain, a DUF283 domain, platform, a PAZ domain, a connector helix, two RNase III domains (RNase IIIa and RNase IIIb) and a dsRNA-binding domain. Specific, ~20-base pair siRNA or miRNA duplexes with 2 nucleotide (nt) 3’-overhangs are generated by Dicer when an RNA substrate is anchored within the platform-PAZ-connector helix (PPC) region. However, increasing number of reports indicate that in the absence of the PAZ domain, binding of RNA substrates can occur by other Dicer domains. Interestingly, truncated variants of Dicer, lacking the PPC region, have been found to display a DNase activity. Inspired by these findings, we investigated how the lack of the PAZ domain, or the entire PPC region, would influence the cleavage activity of hDicer. Using immunopurified 3xFlag-hDicer produced in human cells and its two variants: one lacking the PAZ domain, and the other lacking the entire PPC region, we show that the PAZ domain deletion variants of hDicer are not able to process a pre-miRNA substrate, a dsRNA with 2-nt 30-overhangs, and a blunt-ended dsRNA. -

Genetic Insight Into the Domain Structure and Functions of Dicer-Type Ribonucleases
International Journal of Molecular Sciences Review Genetic Insight into the Domain Structure and Functions of Dicer-Type Ribonucleases Kinga Ciechanowska, Maria Pokornowska and Anna Kurzy ´nska-Kokorniak* Department of Ribonucleoprotein Biochemistry, Institute of Bioorganic Chemistry Polish Academy of Sciences, Noskowskiego 12/14, 61-704 Poznan, Poland; [email protected] (K.C.); [email protected] (M.P.) * Correspondence: [email protected]; Tel.: +48-61-852-85-03 (ext. 1264) Abstract: Ribonuclease Dicer belongs to the family of RNase III endoribonucleases, the enzymes that specifically hydrolyze phosphodiester bonds found in double-stranded regions of RNAs. Dicer enzymes are mostly known for their essential role in the biogenesis of small regulatory RNAs. A typical Dicer-type RNase consists of a helicase domain, a domain of unknown function (DUF283), a PAZ (Piwi-Argonaute-Zwille) domain, two RNase III domains, and a double-stranded RNA binding domain; however, the domain composition of Dicers varies among species. Dicer and its homologues developed only in eukaryotes; nevertheless, the two enzymatic domains of Dicer, helicase and RNase III, display high sequence similarity to their prokaryotic orthologs. Evolutionary studies indicate that a combination of the helicase and RNase III domains in a single protein is a eukaryotic signature and is supposed to be one of the critical events that triggered the consolidation of the eukaryotic RNA interference. In this review, we provide the genetic insight into the domain organization and structure of Dicer proteins found in vertebrate and invertebrate animals, plants and fungi. We also discuss, in the context of the individual domains, domain deletion variants and partner proteins, a variety of Dicers’ functions not only related to small RNA biogenesis pathways. -

The Rnase III Family: a Conserved Structure and Expanding Functions in Eukaryotic Dsrna Metabolism
Curr. Issues Mol. Biol. (2001) 3(4): 71-78. The Eukaryotic RNase III 71 The RNase III Family: A Conserved Structure and Expanding Functions in Eukaryotic dsRNA Metabolism Bruno Lamontagne, Stéphanie Larose, Jim Boulanger, family (Figure 1). In bacteria, RNase III exists in one form and Sherif Abou Elela* characterized by a classical RNA binding domain and a nuclease domain (Nicholson, 1999). In contrast, eukaryotic Département de Microbiologie et d’Infectiologie, Faculté RNase III exists in three isoforms that share the basic de Médecine, Université de Sherbrooke, Sherbrooke, dsRBD but differ in the number of nuclease domains and Québec, Canada J1H 5N4 in the composition of the N-terminal domain (Filippov et al., 2000; Jacobsen et al., 1999; Lamontagne et al., 2000). The first form contains three domains; the dsRBD, the Abstract nuclease domain, and an additional uniquely eukaryotic N-terminal domain required for correct protein conformation The last few years have witnessed the appreciation of and efficient RNA cleavage (Lamontagne et al., 2000). The dsRNA as a regulator of gene expression, a potential second form exhibits in addition to these three domains a antiviral agent, and a tumor suppressor. However, in second nuclease motif at the protein N-terminus (Wu et spite of these clear effects on the cell function, the al., 2000). Finally, the third form of eukaryotic RNase III mechanism that controls dsRNA maturation and contains in addition to the three main eukaryotic domains stability remains unknown. Recently, the discovery of a fourth distinct helicase domain (Jacobsen et al., 1999; eukaryotic orthologues of the bacterial dsRNA specific Rotondo and Frendewey, 1996). -
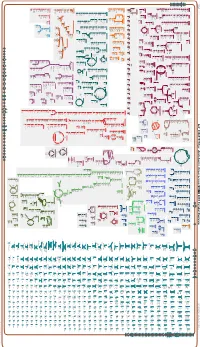
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Peter D Karp Ingrid Keseler An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Anamika Kothari Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000817975Cyc: Azotobacter chroococcum NCIMB 8003 Cellular Overview Connections between pathways are omitted for legibility.