Influence of ARHGAP29 on the Invasion of Mesenchymal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Four-Dimensional Live Imaging of Apical Biosynthetic Trafficking Reveals a Post-Golgi Sorting Role of Apical Endosomal Intermediates
Four-dimensional live imaging of apical biosynthetic trafficking reveals a post-Golgi sorting role of apical endosomal intermediates Roland Thuenauera,b,1,2, Ya-Chu Hsua, Jose Maria Carvajal-Gonzaleza,3, Sylvie Debordea,4, Jen-Zen Chuanga, Winfried Römerc,d, Alois Sonnleitnerb, Enrique Rodriguez-Boulana,5, and Ching-Hwa Sunga,5 aMargaret M. Dyson Vision Research Institute, Weill Medical College of Cornell University, New York, NY 10065; bCenter for Advanced Bioanalysis Linz, 4020 Linz, Austria; and cInstitute of Biology II, and dBIOSS Centre for Biological Signalling Studies, Albert-Ludwigs-University Freiburg, 79104 Freiburg, Germany Edited by Keith E. Mostov, University of California School of Medicine, San Francisco, CA, and accepted by the Editorial Board January 17, 2014 (received for review March 11, 2013) Emerging data suggest that in polarized epithelial cells newly is an important regulator of biological processes that require synthesized apical and basolateral plasma membrane proteins apical trafficking, e.g., lumen formation during epithelial tubu- traffic through different endosomal compartments en route to the logenesis (11), apical secretion of discoidal/fusiform vesicles in respective cell surface. However, direct evidence for trans-endo- bladder umbrella cells (12), and apical microvillus morphogenesis somal pathways of plasma membrane proteins is still missing and and rhodopsin localization in fly photoreceptors (13). However, the mechanisms involved are poorly understood. Here, we imaged despite the physiological importance of trans-endosomal traf- the entire biosynthetic route of rhodopsin-GFP, an apical marker in ficking, the underlying mechanisms remain largely unclear. epithelial cells, synchronized through recombinant conditional ag- Previous studies on trans-endosomal trafficking in polarized gregation domains, in live Madin-Darby canine kidney cells using epithelial cells have relied on pulse chase/cell fractionation pro- spinning disk confocal microscopy. -

Functional Role of the C-Terminal Amphipathic Helix 8 of Olfactory Receptors and Other G Protein-Coupled Receptors
International Journal of Molecular Sciences Review Functional Role of the C-Terminal Amphipathic Helix 8 of Olfactory Receptors and Other G Protein-Coupled Receptors Takaaki Sato 1,*, Takashi Kawasaki 1, Shouhei Mine 1 and Hiroyoshi Matsumura 2 1 Biomedical Research Institute, National Institute of Advanced Industrial Science and Technology, 1-8-31 Midorioka, Ikeda, Osaka 563-8577, Japan; [email protected] (T.K.); [email protected] (S.M.) 2 College of Life Sciences, Ritsumeikan University, Kusatsu, Shiga 525-8577, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-72-751-8342 Academic Editor: Kathleen Van Craenenbroeck Received: 28 September 2016; Accepted: 14 November 2016; Published: 18 November 2016 Abstract: G protein-coupled receptors (GPCRs) transduce various extracellular signals, such as neurotransmitters, hormones, light, and odorous chemicals, into intracellular signals via G protein activation during neurological, cardiovascular, sensory and reproductive signaling. Common and unique features of interactions between GPCRs and specific G proteins are important for structure-based design of drugs in order to treat GPCR-related diseases. Atomic resolution structures of GPCR complexes with G proteins have revealed shared and extensive interactions between the conserved DRY motif and other residues in transmembrane domains 3 (TM3), 5 and 6, and the target G protein C-terminal region. However, the initial interactions formed between GPCRs and their specific G proteins remain unclear. Alanine scanning mutagenesis of the murine olfactory receptor S6 (mOR-S6) indicated that the N-terminal acidic residue of helix 8 of mOR-S6 is responsible for initial transient and specific interactions with chimeric Gα15_olf, resulting in a response that is 2.2-fold more rapid and 1.7-fold more robust than the interaction with Gα15. -

Open Questions: What About the 'Other' Rho Gtpases?
Ridley BMC Biology (2016) 14:64 DOI 10.1186/s12915-016-0289-7 COMMENT Open Access Open questions: what about the ‘other’ Rho GTPases? Anne J. Ridley Abstract know if they interact with and/or regulate the activity of other family members. Indeed, by studying only RhoA, Rho GTPases have many and diverse roles in cell Rac1 and Cdc42, we are likely to be missing the real physiology, and some family members are very well functions of many GEFs and GAPs because their targets studied, including RhoA, Rac1 and Cdc42. But many in cells are among the other Rho GTPases. are relatively neglected, and fundamental questions Interestingly, four family members—Rnd1, Rnd2, Rnd3 about their mechanisms and functions remain open. and RhoH—are ‘atypical’, in that they are known to be constitutively GTP-bound and do not hydrolyse GTP: much less is known about how these family members Rho GTPases are household names for anyone who are regulated. RhoU and RhoV have high intrinsic GDP/ works on eukaryotic cell migration and their functions GTP exchange rates, so are unlikely to need GEFs for in cell migration, cell division and cell polarity are de- activation but could still be turned off by GAPs [1, 4]. scribed in most textbooks on cell biology. Yet most of For two other members, RhoBTB1 and RhoBTB2, the what we know about Rho GTPases comes from studying Rho domains are quite divergent in sequence from other a small subset of the many different family members, family members and they are unlikely to be regulated by namely RhoA, Rac1 and Cdc42. -

(12) United States Patent (10) Patent No.: US 9,695.227 B2 Steyaert Et Al
USOO9695227B2 (12) United States Patent (10) Patent No.: US 9,695.227 B2 Steyaert et al. (45) Date of Patent: Jul. 4, 2017 (54) BINDING DOMAINS DIRECTED AGAINST (58) Field of Classification Search GPCRG PROTEIN COMPLEXES AND USES None DERVED THEREOF See application file for complete search history. (56) References Cited (75) Inventors: Jan Steyaert, Beersel (BE); Els Pardon, Lubbeek (BE); Toon U.S. PATENT DOCUMENTS Laeremans, Dworp (BE); Brian 5,721,121 A 2/1998 Etcheverry et al. Kobilka, Palo Alto, CA (US): Soren 2007/OO77597 A1 4/2007 Gilchrist et al. Rasmussen, Frederiksberg (DK); 2007/0231830 A1 10/2007 Gilchrist et al. Sebastian Granier, Menlo Park, CA (US); Roger K. Sunahara, Ann Arbor, FOREIGN PATENT DOCUMENTS MI (US) WO 94.04678 A1 3, 1994 WO 993.7681 A2 7, 1999 (73) Assignees: Vrije Universiteit Brussel, Brussels WO OO43507 A2 T 2000 (BE); VIB VZW, Ghent (BE); The WO O190190 A2 11/2001 Board of Trustees of the Leland WO O2O85945 A2 10, 2002 Stanford Junior Universitv, Palo Alto, WO O3O25O2O A1 3, 2003 CA (US); The Regents of the WO O3035694 A2 5, 2003 WO 2004035614 A1 4/2004 University of Michigan, Ann Arbor, WO 2004.049.794 A2 6, 2004 MI (US) WO 2006086883 A1 8, 2006 WO 2009051633 4/2009 (*) Notice: Subject to any disclaimer, the term of this WO 2009,147.196 A1 12/2009 patent is extended or adjusted under 35 WO 2010043650 A2 4/2010 WO 201OO6674.0 A1 6, 2010 U.S.C. 154(b) by 594 days. WO 2012007593 A1 1, 2012 (21) Appl. -

Itga4 Cldn16 Cldn9 Cldn15 Cldn22 Ocln Esam
Supplementary material J Med Genet Table S1. List of 263 genes included in the AGS-LEUK panel. Axonal Guidance Signaling genes as AGS and Leukocyte transvasation genes as LEUK. List of genes (AGS) List of genes (LEUK) ABLIM1 CLDN11 ACTR3 MMP14 ADAM11 MMP15 ADAM23 CTNNA1 ADAMTS1 ENSG00000130396 ADAMTS4 CLDN6 ADAMTS9 MMP24 ARHGEF15 ARHGAP12 ARHGEF6 DLC1 ARPC1B TIMP2 BDNF RAPGEF3 BMP1 F11R BMP4 CLDN23 BMP6 CLDN8 BMP7 JAM3 CXCL12 CLDN3 CXCR4 ARHGAP8 DPYSL5 ICAM1 EFNA1 MMP16 EFNA5 JAM2 ENPEP CLDN7 EPHA1 TIMP3 EPHA3 VCAM1 EPHA5 CLDN5 EPHA7 MSN EPHB1 NOX3 EPHB2 ACTC1 EPHB4 VAV2 FGFR2 CLDN10 FZD1 RAP1GAP FZD10 VAV3 FZD5 MAPK10 FZD6 CTNNA2 GAB1 CDH5 GLI1 PECAM1 GLI3 CTNND1 GNA14 ITGA4 GNAI1 CLDN16 GNAO1 CLDN9 GNAS CLDN15 GNB4 CLDN22 GNG11 OCLN GNG2 ESAM Gallego-Martinez A, et al. J Med Genet 2019; 0:1–7. doi: 10.1136/jmedgenet-2019-106159 Supplementary material J Med Genet GNG7 ACTB IGF1 CYBA IRS1 CTNNB1 IRS2 MMP9 ITGA3 MAPK14 ITGB1 MAPK11 LIMK1 MAPK12 LIMK2 MAPK13 LINGO1 PRKCB LRRC4C PXN MME BCAR1 MMP11 THY1 MMP2 ARHGAP5 MRAS MYL2 MYL9 MYLPF NFATC4 RAP1A NGFR RAP1B NOTUM VASP NRP1 ACTN4 NTN3 ACTN1 NTRK2 VCL NTRK3 RAPGEF4 PAK3 ITK PAK4 VAV1 PAPPA2 PDGFA PDGFC PIK3CB PIK3R1 PLCE1 PLCH1 PLCH2 PLXNA2 PLXNB1 PLXND1 PPP3CA PRKACB PRKAR2A PRKAR2B PRKCA PRKCZ PRKD3 ROBO2 SDC2 SDCBP Gallego-Martinez A, et al. J Med Genet 2019; 0:1–7. doi: 10.1136/jmedgenet-2019-106159 Supplementary material J Med Genet SEMA3B SEMA3C SEMA3E SEMA3F SEMA4F SEMA4G SEMA5A SEMA6B SEMA6D SEMA7A SHC1 SLIT2 SLIT3 STK36 TUBA4A TUBB2B TUBB4A TUBB4B TUBB6 UNC5C UNC5D ENSG00000165197 WIPF1 WNT3 WNT5A WNT7A WNT7B NTNG1 NTNG2 LRRC4 NTN4 TRPC1 TRPC3 TRPC6 PPP3CB PPP3CC PPP3R1 NFATC2 NFATC3 PTK2 FYN RAC1 CDC42 ABLIM2 NCK1 PAK1 PAK2 Gallego-Martinez A, et al. -

Bacterial Toxins That Target Rho Proteins
Bacterial toxins that target Rho proteins. K Aktories J Clin Invest. 1997;99(5):827-829. https://doi.org/10.1172/JCI119245. Perspective Find the latest version: https://jci.me/119245/pdf Perspectives Series: Host/Pathogen Interactions Bacterial Toxins That Target Rho Proteins Klaus Aktories Institut für Pharmakologie and Toxikologie, Albert-Ludwigs-Universität Freiburg, D-79104 Freiburg i.Br., Germany Not only “big” GTP-binding proteins (heterotrimeric G-pro- ular switches in various signal transduction processes, e.g., Rho teins, elongation factor 2) but also “small” GTPases are targets subfamily proteins participate in control of cell–cell contact, for bacterial protein toxins. In this respect, Rho subfamily pro- integrin signaling, endocytosis, transcriptional activation, pro- teins are of particular importance. These GTPases are ADP- liferation, apoptosis, and cell transformation. Accordingly, nu- ribosylated by Clostridium botulinum C3-like transferases and merous potential Rho-interacting effectors have been de- are monoglycosylated by large clostridial cytotoxins. They ap- scribed, some of which are kinases (e.g., Rho kinase, p60 PAK pear to be the target of Escherichia coli cytotoxic necrotizing kinase, PKN kinase, lipid kinases), and some of which are factors (CNF1, 2)1 and Bordetella dermonecrotic toxin (DNT). adaptor proteins without kinase activity (e.g., WASP, p67 Moreover, Rho proteins seem to be essential for uptake of phox). Several of these processes appear to be important for bacteria into eukaryotic cells. bacteria–host interactions, signal transduction of cells of the immune system, and host defense mechanisms. This is particu- Rho proteins larly evident in the case of Rac and its interaction with Rho proteins (Rho stands for Ras homologous proteins) be- p67phox, which is an essential cofactor for superoxide anion long to the superfamny of Ras proteins (1). -
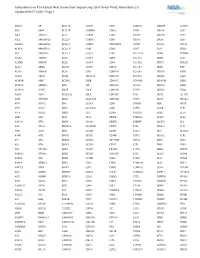
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3 -

Supplementary Data
Supplementary Figure 1 Supplementary Figure 2 CCR-10-3244.R1 Supplementary Figure Legends Supplementary Figure 1. B-Myb is overexpressed in primary AML blasts and B-CLL cells. Baseline B-Myb mRNA levels were determined by quantitative RT-PCR, after normalization to the level of housekeeping gene, in primary B-CLL (n=10) and AML (n=5) patient samples, and in normal CD19+ (n=5) and CD34+ (n=4) cell preparations. Each sample was determined in triplicate. Horizontal bars are median, upper and lower edges of box are 75th and 25th percentiles, lines extending from box are 10th and 90th percentiles. Supplementary Figure 2. Cytotoxicity by Nutlin-3 and Chlorambucil used alone or in combination in leukemic cells. The p53wild-type EHEB and SKW6.4 cells lines, and the p53mutated BJAB cell line were exposed to Nutlin-3 or Chlorambucil used either alone or in combination. (Nutl.+Chlor.). In A, upon treatment with Nutlin-3 or Chlorambucil, used either alone (both at 10 μM) or in combination (Nutl.+Chlor.), induction of apoptosis was quantitatively evaluated by Annexin V/PI staining, while E2F1 and pRb protein levels were analyzed by Western blot. Tubulin staining is shown as loading control. The average combination index (CI) values (analyzed by the method of Chou and Talalay) for effects of Chlorambucil+Nutlin-3 on cell viability are shown. ED indicates effect dose. In B, levels of B-Myb and E2F1 mRNA were analyzed by quantitative RT- PCR. Results are expressed as fold of B-Myb and E2F1 modulation in cells treated for 24 hours as indicated, with respect to the control untreated cultures set to 1 (hatched line). -

Rnd3-Induced Cell Rounding Requires Interaction with Plexin-B2 Brad Mccoll*,¶, Ritu Garg¶, Philippe Riou‡, Kirsi Riento§ and Anne J
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 4046-4056 doi:10.1242/jcs.192211 RESEARCH ARTICLE Rnd3-induced cell rounding requires interaction with Plexin-B2 Brad McColl*,¶, Ritu Garg¶, Philippe Riou‡, Kirsi Riento§ and Anne J. Ridley** ABSTRACT migration during mouse development, in part because their Rnd proteins are atypical members of the Rho GTPase family that expression is regulated by different transcription factors (Pacary induce actin cytoskeletal reorganization and cell rounding. Rnd et al., 2011). Interestingly, in endothelial cells, both Rnd2 and Rnd3 proteins have been reported to bind to the intracellular domain of induce stress fibre assembly by upregulating RhoB expression several plexin receptors, but whether plexins contribute to the Rnd- (Gottesbuhren et al., 2013). induced rounding response is not known. Here we show that Rnd3 Several proteins that interact with Rnd proteins and could mediate interacts preferentially with plexin-B2 of the three plexin-B proteins, their effects on cell shape and motility have been reported (Riou whereas Rnd2 interacts with all three B-type plexins, and Rnd1 et al., 2010). The first to be identified was p190RhoGAP, which shows only very weak interaction with plexin-B proteins in downregulates RhoA activity. Rnd3 interaction with p190RhoGAP immunoprecipitations. Plexin-B1 has been reported to act as a GAP stimulates its activity and induces loss of stress fibres (Wennerberg for R-Ras and/or Rap1 proteins. We show that all three plexin-B et al., 2003). Rnd3, but not Rnd1 or Rnd2, also binds to and is proteins interact with R-Ras and Rap1, but Rnd proteins do not alter phosphorylated by the serine/threonine kinases ROCK1 and PKC this interaction or R-Ras or Rap1 activity. -

The RHO Family Gtpases: Mechanisms of Regulation and Signaling
cells Review The RHO Family GTPases: Mechanisms of Regulation and Signaling Niloufar Mosaddeghzadeh and Mohammad Reza Ahmadian * Institute of Biochemistry and Molecular Biology II, Medical Faculty of the Heinrich Heine University, Universitätsstrasse 1, Building 22.03.05, 40225 Düsseldorf, Germany; [email protected] * Correspondence: [email protected] Abstract: Much progress has been made toward deciphering RHO GTPase functions, and many studies have convincingly demonstrated that altered signal transduction through RHO GTPases is a recurring theme in the progression of human malignancies. It seems that 20 canonical RHO GTPases are likely regulated by three GDIs, 85 GEFs, and 66 GAPs, and eventually interact with >70 downstream effectors. A recurring theme is the challenge in understanding the molecular determinants of the specificity of these four classes of interacting proteins that, irrespective of their functions, bind to common sites on the surface of RHO GTPases. Identified and structurally verified hotspots as functional determinants specific to RHO GTPase regulation by GDIs, GEFs, and GAPs as well as signaling through effectors are presented, and challenges and future perspectives are discussed. Keywords: CDC42; effectors; RAC1; RHOA; RHOGAP; RHOGDI; RHOGEF; RHO signaling 1. Introduction Citation: Mosaddeghzadeh, N.; The RHO (RAS homolog) family is an integral part of the RAS superfamily of guanine Ahmadian, M.R. The RHO Family nucleotide-binding proteins. RHO family proteins are crucial for several reasons: (i) ap- GTPases: Mechanisms of Regulation proximately 1% of the human genome encodes proteins that either regulate or are regulated and Signaling. Cells 2021, 10, 1831. by direct interaction with RHO proteins; (ii) they control almost all fundamental cellular https://doi.org/10.3390/cells10071831 processes in eukaryotes including morphogenesis, polarity, movement, cell division, gene expression, and cytoskeleton reorganization [1]; and (iii) they are associated with a series Academic Editor: Bor Luen Tang of human diseases (Figure1)[2]. -
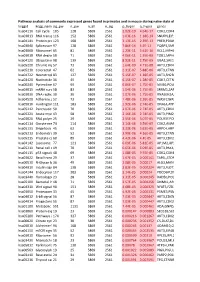
Pathway Analysis of Commonly Expressed Genes Found in Primates and in Mouse During Naïve State of Pluripotenc Keggid Kegg Names
Pathway analysis of commonly expressed genes found in primates and in mouse during naïve state of pluripotency. keggid kegg_namesig_pw n_pw n_all n_sig p_hyper q_hyper genes hsa04110 Cell cycle 105 128 5869 2561 2.02E-19 4.64E-17 CDK2,CDK4,CDK6,CDK7,CDKN1A,CDKN1B,STAG1,CDKN2B,ANAPC10,MAD2L2,STAG2,GADD45G,DBF4,YWHAQ,CHEK1,CHEK2,CREBBP,GADD45A,E2F1,E2F3,E2F4,E2F5,EP300,ORC3,CDC26,ABL1,ANAPC13,SFN,GSK3B,ANAPC2,ANAPC4,HDAC1,HDAC2,MAD2L1,SMAD2,SMAD3,SMAD4,MCM2,MCM3,MCM4,MCM5,MCM6,MCM7,MDM2,MYC,GADD45B,ATM,ORC1,ORC2,ORC4,ORC5,FZR1,ANAPC7,ANAPC11,PLK1,ATR,PRKDC,RAD21,RB1,RBL1,CCND1,ANAPC1,SKP2,BUB1,BUB1B,TFDP1,TFDP2,TGFB1,TGFB2,TTK,WEE1,YWHAB,YWHAE,YWHAG,YWHAH,YWHAZ,ZBTB17,SMC1A,CDC7,CDC45,MAD1L1,CDC14A,CDC23,CDC16,CCNA2,CCNB1,CCND2,CCND3,CCNE1,CCNH,PKMYT1,SMC3,CCNB2,CCNE2,BUB3,PTTG1,ESPL1,CDK1,CDC6,CDC20,CDC25A,CDC25B,CDC25C,CDC27,RBX1 hsa03013 RNA transport116 152 5869 2561 1.03E-16 1.18E-14 SNUPN,EIF1,POP7,SRRM1,SAP18,EIF1B,PRMT5,TACC3,NXF1,RPP30,RPP38,PAIP1,POP4,RPP40,RNPS1,POP1,STRAP,DDX20,XPOT,CLNS1A,NUP35,RPP25L,EEF1A1,EEF1A2,EIF2S1,EIF2B1,EIF4A1,EIF4B,EIF4E,EIF4EBP1,EIF4G1,EIF4G2,EIF5,CASC3,NCBP2,ACIN1,NUP205,NUP210,NUP62,GEMIN5,UPF2,PABPC1,NXT1,NUP43,EIF3E,KPNB1,MAGOH,NCBP1,NUP88,NUP98,GEMIN4,NMD3,POP5,PHAX,NUP54,PNN,RPP25,GEMIN8,MAGOHB,ELAC1,NDC1,NUP133,NXT2,NUP107,THOC2,XPO5,RAN,RANBP2,RANGAP1,SENP2,UPF1,ELAC2,SEC13,UPF3B,SMN1,SUMO3,SUMO2,TPR,SUMO1,XPO1,NUP37,DDX39B,THOC6,GEMIN7,GEMIN6,RPP21,NUP85,THOC7,NUP214,AAAS,SEH1L,THOC3,RAE1,THOC5,EIF3A,EIF3B,EIF3C,EIF3D,EIF3F,EIF3G,EIF3H,EIF3I,EIF4G3,PABPC4,EIF2B4,EIF2B2,EIF2B5,EIF2S2,EIF4E2,NUP155,EIF5B,TGS1,NUP93,EIF4A3,NUP153,THOC1