Separation of Formic Acid/3-Methyl-2-Butanone, Formic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methyl Ethyl Ketone (2-Butanone)
Methyl Ethyl Ketone (2-Butanone) 78-93-3 Hazard Summary Methyl ethyl ketone is used as a solvent. Acute (short-term) inhalation exposure to methyl ethyl ketone in humans results in irritation to the eyes, nose, and throat. Limited information is available on the chronic (long-term) effects of methyl ethyl ketone in humans. Chronic inhalation studies in animals have reported slight neurological, liver, kidney, and respiratory effects. No information is available on the developmental, reproductive, or carcinogenic effects of methyl ethyl ketone in humans. Developmental effects, including decreased fetal weight and fetal malformations, have been reported in mice and rats exposed to methyl ethyl ketone via inhalation and ingestion. EPA has classified methyl ethyl ketone as a Group D, not classifiable as to human carcinogenicity. Please Note: The main sources of information for this fact sheet are EPA's Health Effects Assessment for Methyl Ethyl Ketone (1) and EPA's Integrated Risk Information System (IRIS) (6), which contains information on inhalation chronic toxicity of methyl ethyl ketone and the RfC and oral chronic toxicity and the RfD. Uses The primary use of methyl ethyl ketone is as a solvent in processes involving gums, resins, cellulose acetate, and cellulose nitrate. (1) Methyl ethyl ketone is also used in the synthetic rubber industry, in the production of paraffin wax, and in household products such as lacquer and varnishes, paint remover, and glues. (1) Sources and Potential Exposure Methyl ethyl ketone has been detected in both indoor and outdoor air. Methyl ethyl ketone can be produced in outdoor air by the photooxidation of certain air pollutants, such as butane and other hydrocarbons. -

Methyl Isopropyl Ketone Safety Data Sheet According to Federal Register / Vol
Methyl isopropyl ketone Safety Data Sheet according to Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules and Regulations Date of issue: 05/25/2015 Version: 1.0 SECTION 1: Identification 1.1. Identification Product form : Substance Substance name : Methyl isopropyl ketone CAS-No. : 563-80-4 Product code : (US) W1814 Formula : C5H10O Synonyms : 3-Methylbutane-2-one / Isopropyl methyl ketone / Methyl-2-butanone, 3- / 2-Acetylpropane / 3- Methylbutanone-2 / 3-Methylbutanone / 3-Methylbutan-2-one / 3-Methyl-2-butanone / 2- Butanone, 3-methyl- / Butan-2-one, 3-methyl- 1.2. Recommended use and restrictions on use No additional information available 1.3. S upplie r Synerzine 5340 Hwy 42 S Ellenwood, Georgia 30294 - USA T 404-524-6744 - F 404-577-1651 [email protected] - www.synerzine.com 1.4. Emergency telephone number Emergency number : Infotrac 1-800-535-5053 (Contract# 102471) Dial +1-352-323-3500 when outside the US SECTION 2: Hazard(s) identification 2.1. Classification of the substance or mi xt ure GHS -US classification Flammable liquids Category H225 Highly flammable liquid and vapour 2 Specific target organ toxicity H336 May cause drowsiness or dizziness (single exposure) Category 3 Hazardous to the aquatic H402 Harmful to aquatic life environment - Acute Hazard Category 3 Full text of H statements : see section 16 2.2. GHS Label elements, including precautionary statements GHS-US labeling Hazard pictograms (GHS-US) : Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H225 - Highly flammable liquid and vapour H336 - May cause drowsiness or dizziness H402 - Harmful to aquatic life Precautionary statements (GHS-US) : P210 - Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. -

Toxicological Profile for 2-Butanone Released for Public Comment in May 2019
Toxicological Profile for 2-Butanone October 2020 2-BUTANONE ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. 2-BUTANONE iii FOREWORD This toxicological profile is prepared in accordance with guidelines* developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. The adequacy of information to determine a substance's -

References on Use of CO2 for Medical
Some References on the Use of CO2 for Medical Entomology Survey Becker N et al. Comparison of carbon dioxide, octenol and a host-odour as mosquito attractants in the Upper Rhine Valley, Germany. Med Vet Entomol 1995;9:377-80. Abstract: Field studies were conducted in the Upper Rhine Valley to determine the responses of mosquitoes to CDC traps baited with either CO2, octenol, light or paired combinations of these. Among eight mosquito species caught, the attractant effect on trap catches was studied in the four most abundant: Aedes vexans, Ae. rossicus, Ae. cinereus and Culex pipiens. Traps baited only with light or octenol caught few mosquitoes, whereas many were caught by traps baited with CO2 alone or in combination with either of the other candidate attractants. CO2 baited traps, with or without light, caught the most Aedes. The combination of CO2 and octenol attracted most Cx pipiens, but this apparent synergy was not significant. Using a caged hamster compared to CO2 as bait in a CDC light-trap with only intermittent fan suction, the hamster attracted less mosquitoes than CO2 emitted at a rate of 225 g/h on days 1 and 2, whereas on days 3 and 4 the smell from the hamster's cage became significantly more attractive than this rate of CO2 for all species of mosquitoes. Canyon DV, Hii JL. Efficacy of carbon dioxide, 1-octen-3-ol, and lactic acid in modified Fay- Prince traps as compared to man-landing catch of Aedes aegypti. J Am Mosq Control Assoc 1997;13:66-70. Abstract: The attractants 1-octen-3-ol and lactic acid significantly decreased catches of Aedes aegypti in Townsville, Australia, by 50% in a controlled laboratory environment and by 100% in the field when compared to carbon dioxide baited bidirectional Fay-Prince trap catches. -

Synthesis of Polyesters by the Reaction of Dicarboxylic Acids with Alkyl Dihalides Using the DBU Method
Polymer Journal, Vol. 22, No. 12, pp 1043-1050 (1990) Synthesis of Polyesters by the Reaction of Dicarboxylic Acids with Alkyl Dihalides Using the DBU Method Tadatomi NISHIKUBO* and Kazuhiro OZAKI Department of Applied Chemistry, Faculty of Engineering, Kanagawa University, Rokkakubashi, Kanagawa-ku, Yokohama 221, Japan (Received July 6, 1990) ABSTRACT: Some polyesters with moderate viscosity were synthesized by reactions of dicarboxylic acids with alkyl dihalides using 1,8-diazabicyclo-[5.4.0]-7-undecene (DBU) in aprotic polar solvents such as dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) under relatively mild conditions. The viscosity and yield of the resulting polymer increased with increasing monomer concentration. Although polymers with relatively high viscosity were obtained when the reaction with p-xylylene dichloride was carried out at 70°C in DMSO, the viscosity of the resulting polymers decreased with increasing reaction temperature when the reaction with m-xylylene dibromide was carried out in DMSO. KEY WORDS Polyester Synthesis/ Dicarboxylic Acids/ Alkyl Dihalides / DBU Method / Mild Reaction Condition / Although poly(ethylene terephthalate) is favorable method for the synthesis of polyes synthesized industrially by transesterification ters because the preparation and purification between dimethyl terephthalate and ethylene of the activated. dicarboxylic acids is un glycol at relatively high temperatures using necessary. certain catalysts, many polyesters are usually Some polyesters have also been prepared8 prepared by the polycondensation of dicarbox by reactions between alkali metal salts of ylic-acid chlorides with difunctional alcohols dicarboxylic-acids and aliphatic dibromides or phenols. These reactions are carried out using phase transfer catalysis (PTC)s, which is under relatively mild conditions; however, the a very convenient method for chemical activated dicarboxylic-acid chlorides must be modification, especially esterification9 or ether prepared and purified before the reaction. -

Liquid Phase Oxidation of P-Xylene to Terephthalic Acid at Medium-High Temperatures: Multiple Benefits of CO2-Expanded Liquids
View Article Online / Journal Homepage / Table of Contents for this issue PAPER www.rsc.org/greenchem | Green Chemistry Liquid phase oxidation of p-xylene to terephthalic acid at medium-high temperatures: multiple benefits of CO2-expanded liquids Xiaobin Zuo,a,b Fenghui Niu,a Kirk Snavely,a,c Bala Subramaniam*a,c and Daryle H. Busch*a,b Received 29th September 2009, Accepted 18th November 2009 First published as an Advance Article on the web 15th January 2010 DOI: 10.1039/b920262e The Co/Mn/Br catalyzed oxidation of p-xylene to terephthalic acid (TPA) is demonstrated in CO2-expanded solvents at temperatures lower than those of the traditional Mid-Century (MC) process. As compared with the traditional air (N2/O2) oxidation system, the reaction with CO2/O2 ◦ mixture at 160 C and using an additional inert gas (N2 or CO2) pressure of 100 bar increases both the yield of TPA and the purity of solid TPA via a more efficient conversion of the intermediates, 4-carboxybenzaldehyde and p-toluic acid. At the same time, the amount of yellow colored by-products in the solid TPA product is also lessened, as determined by spectroscopic analysis. Equally important, the decomposition or burning of the solvent, acetic acid, monitored in terms of the yield of the gaseous products, CO and CO2, is reduced by ca.20%basedonlabeledCO2 experiments. These findings broaden the versatility of this new class of reaction media in homogeneous catalytic oxidations by maximizing the utilization of feedstock carbon for desired products while simultaneously reducing carbon emissions. 1. Introduction limited by the even harsher reaction conditions (T = 300–400 ◦C, P > 200 bar). -

Dose–Response Assay for Synthetic Mosquito (Diptera: Culicidae) Attractant Using a High-Throughput Screening System
insects Article Dose–Response Assay for Synthetic Mosquito (Diptera: Culicidae) Attractant Using a High-Throughput Screening System Dae-Yun Kim 1, Theerachart Leepasert 2, Michael J. Bangs 1 and Theeraphap Chareonviriyaphap 1,* 1 Department of Entomology, Faculty of Agriculture, Kasetsart Univeristy, Bangkok 10900, Thailand; [email protected] (D.-Y.K.); [email protected] (M.J.B.) 2 Department of Chemistry, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand; [email protected] * Correspondence: [email protected] Simple Summary: Entomological surveillance is important to evaluate vector management inter- ventions. However, collecting adult mosquitoes using direct human bait is controversial and often discouraged because of potential infection risk. Alternatively, active and passive trapping methods are available. Female mosquitoes detect human host cues such as body heat, carbon dioxide, and other volatile body emanations using olfactory sensilla to direct movement to a host. Attractive chemical lures have been identified and evaluated using a variety of olfactometric methods to in- crease trap production and efficiency. In this study, we evaluated a simple olfactometer without need of airflow. To ‘optimize’ a commercial mosquito attractant, 10 different doses of product, the TM Biogents-lure (BG-lure ), were compared. Results showed dose-dependent responses with 0.005 g with the highest attraction for Aedes aegypti, while doses of 0.2 g and above produced a repellent Citation: Kim, D.-Y.; Leepasert, T.; response. There was no significantly different response behavior between permethrin-susceptible Bangs, M.J.; Chareonviriyaphap, T. and -resistant Ae. aegypti. Culex quinquefasciatus showed significantly different responses compared Dose–Response Assay for Synthetic to Ae. -
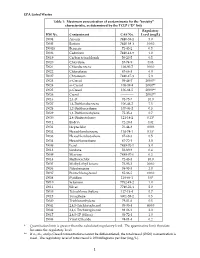
EPA Listed Wastes Table 1: Maximum Concentration of Contaminants For
EPA Listed Wastes Table 1: Maximum concentration of contaminants for the toxicity characteristic, as determined by the TCLP (D list) Regulatory HW No. Contaminant CAS No. Level (mg/L) D004 Arsenic 7440-38-2 5.0 D005 Barium 7440-39-3 100.0 D0018 Benzene 71-43-2 0.5 D006 Cadmium 7440-43-9 1.0 D019 Carbon tetrachloride 56-23-5 0.5 D020 Chlordane 57-74-9 0.03 D021 Chlorobenzene 108-90-7 100.0 D022 Chloroform 67-66-3 6.0 D007 Chromium 7440-47-3 5.0 D023 o-Cresol 95-48-7 200.0** D024 m-Cresol 108-39-4 200.0** D025 p-Cresol 106-44-5 200.0** D026 Cresol ------------ 200.0** D016 2,4-D 94-75-7 10.0 D027 1,4-Dichlorobenzene 106-46-7 7.5 D028 1,2-Dichloroethane 107-06-2 0.5 D029 1,1-Dichloroethylene 75-35-4 0.7 D030 2,4-Dinitrotoluene 121-14-2 0.13* D012 Endrin 72-20-8 0.02 D031 Heptachlor 76-44-8 0.008 D032 Hexachlorobenzene 118-74-1 0.13* D033 Hexachlorobutadiene 87-68-3 0.5 D034 Hexachloroethane 67-72-1 3.0 D008 Lead 7439-92-1 5.0 D013 Lindane 58-89-9 0.4 D009 Mercury 7439-97-6 0.2 D014 Methoxychlor 72-43-5 10.0 D035 Methyl ethyl ketone 78-93-3 200.0 D036 Nitrobenzene 98-95-3 2.0 D037 Pentachlorophenol 87-86-5 100.0 D038 Pyridine 110-86-1 5.0* D010 Selenium 7782-49-2 1.0 D011 Silver 7740-22-4 5.0 D039 Tetrachloroethylene 127-18-4 0.7 D015 Toxaphene 8001-35-2 0.5 D040 Trichloroethylene 79-01-6 0.5 D041 2,4,5-Trichlorophenol 95-95-4 400.0 D042 2,4,6-Trichlorophenol 88-06-2 2.0 D017 2,4,5-TP (Silvex) 93-72-1 1.0 D043 Vinyl Chloride 74-01-4 0.2 * Quantitation limit is greater than the calculated regulatory level. -

Aging Behavior and Modeling Studies of Unsaturated Polyester Resin and Unsaturated Polyester Resin-Based Blends Emmanuel Richaud, Jacques Verdu
Aging behavior and modeling studies of unsaturated polyester resin and unsaturated polyester resin-based blends Emmanuel Richaud, Jacques Verdu To cite this version: Emmanuel Richaud, Jacques Verdu. Aging behavior and modeling studies of unsaturated polyester resin and unsaturated polyester resin-based blends. Unsaturated Polyester Resins : Fundamentals, Design, Fabrication, and Applications, Elsevier, pp.199-231, 2019, 978-012816129-6. 10.1016/B978- 0-12-816129-6.00009-0. hal-02568819 HAL Id: hal-02568819 https://hal.archives-ouvertes.fr/hal-02568819 Submitted on 10 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. AGING BEHAVIOR AND MODELING STUDIES OF UNSATURATED POLYESTER RESIN AND UNSATURATED POLYESTER RESIN-BASED BLENDS Emmanuel Richaud and Jacques Verdu PIMM, UMR 8006, ENSAM À CNRS À CNAM, HESAM Universite,´ Paris, France 9.1 INTRODUCTION Unsaturated polyester resins (UPRs) are usually obtained from a prepolymer being a condensation product of unsaturated anhydride (or diacid) and a diol. This highly viscous liquid is dissolved in a reactive low viscosity solvent, commonly styrene (or sometimes methyl methacrylate). The curing is initiated by peroxide and metallic salt catalyst of the peroxide decomposition often called “accelerator”. -

Study on the Structure Activity Relationship of the Crystal MOF-5 Synthesis, Thermal Stability and N2 Adsorption Property
High Temp. Mater. Proc. 2020; 39:171–177 Research Article Sheng Wang*, Xianfei Xie, Wenke Xia, Jiaming Cui, Shengquan Zhang, and Xueyan Du Study on the structure activity relationship of the crystal MOF-5 synthesis, thermal stability and N2 adsorption property https://doi.org/10.1515/htmp-2020-0034 storage, chemical sensing, sensors, battery, catalysis, light Received Oct 13, 2018; accepted Mar 04, 2019 to electrical energy conversion, membrane, and even drug delivery due to their unsurpassed porosities, well-defined Abstract: The parallel flow drop solvothermal method was pore structures, high specific surface area, pore volume, utilized to synthesize the crystal of MOF-5 by taking the mo- designable and flexible structure, and strong physical- lar ratio of the metal ions to the organic ligands of 2:1 at chemical stability [1–3]. One of the most widely researched 140∘C, and the reaction time at 12 hours. Meanwhile, the MOFs materials is MOF-5, which has high gas selectiv- structure and properties of MOF-5 were characterized by ity and capacity [4]. MOF-5, namely [Zn O(BDC) ], is a the X-ray diffraction (XRD), scanning electron microscope 4 3 three-dimensional cubic porous framework with [Zn O]6+ (SEM), thermogravimetric analysis (TGA) and fourier trans- 4 clusters linked together through 1,4-benzenedicarboxylate form infrared spectroscopy (FTIR). SEM analysis shown (BDC2−) ligands [5]. Therefore, this specific structure that the crystal morphology of MOF-5 changed from sheet makes MOF-5 large surface area and exceptional pore vol- to cubic with increasing reaction temperature and molar ume. It has rapidly developed as a hotspot in the crossing ratio of the metal ions to the organic ligands, and its ther- fields of energy, chemistry, materials and life science [6]. -

AP-42, CH 6.11: Terephthalic Acid
6.11 Terephthalic Acid 6.11.1 Process Description1 Terephthalic acid (TPA) is made by air oxidation of p-xylene and requires purification for use in polyester fiber manufacture. A typical continuous process for the manufacture of crude terephthalic acid (C-TPA) is shown in Figure 6.11-1. The oxidation and product recovery portion essentially consists of the Mid-Century oxidation process, whereas the recovery and recycle of acetic acid and recovery of methyl acetate are essentially as practiced by dimethyl terephthalate (DMT) technology. The purpose of the DMT process is to convert the terephthalic acid contained in C-TPA to a form that will permit its separation from impurities. C-TPA is extremely insoluble in both water and most common organic solvents. Additionally, it does not melt, it sublimes. Some products of partial oxidation of p-xylene, such as p-toluic acid and p-formyl benzoic acid, appear as impurities in TPA. Methyl acetate is also formed in significant amounts in the reaction. 6.11.1.1 C-TPA Production - Oxidation Of p-Xylene - p-Xylene (stream 1 of Figure 6.11-1), fresh acetic acid (2), a catalyst system such as manganese or cobalt acetate and sodium bromide (3), and recovered acetic acid are combined into the liquid feed entering the reactor (5). Air (6), compressed to a reaction pressure of about 2000 kPa (290 psi), is fed to the reactor. The temperature of the exothermic reaction is maintained at about 200°C (392°F) by controlling the pressure at which the reaction mixture is permitted to boil and form the vapor stream leaving the reactor (7). -

Methyl Ethyl Ketone
Shell Chemicals Technical Datasheet Methyl Ethyl Ketone Product Code S2113 Region Global Product Category Ketones CAS Registry Number 78-93-3 Synonym(s) 2-Butanone, MEK Description Methyl ethyl ketone, MEK, a low boiling, fast evaporating solvent is a Methyl ethyl ketone, MEK, is a low boiling, fast evaporating solvent colourless, stable liquid, partially miscible with water. MEK has exceptionally good solvency for many synthetic and natural resins that are used in the formulation of printing inks, lacquers, and other types of coatings. Typical Properties Property Unit Method Value Purity, min. %m/m GC 99.5 Water %m/m ASTM D1364 0.03 Acidity (as Acetic Acid) %m/m ASTM D1613 0.002 Density at 20°C kg/l ASTM D4052 0.805 Specific Gravity at 20°C/20°C - ASTM D4052 0.806 Specific Gravity at 25°C/25°C - ASTM D4052 0.802 Coefficient of Cubic Expansion at 20°C 10-4/°C Calculated 13 Refractive Index at 20°C - ASTM D1218 1.379 Colour Pt-Co ASTM D1209 < 5 Boiling Point °C - 80 Relative Evaporation Rate (nBuAc=1) - ASTM D3539 4.0 Relative Evaporation Rate (Ether=1) - DIN 53170 3.3 Antoine Constant A # kPa. °C - 6.18444 Antoine Constant B # kPa. °C - 1259.22 Antoine Constant C # kPa. °C - 221.758 26 Methyl Ethyl Ketone March 2016 Temperature Limits for Antoine Equation # °C - -40 to +90 Vapour Pressure at 20°C kPa Calculated 9.5 Vapour Pressure at 50°C kPa Calculated 36 Saturated Vapor Concentration at 20°C g/m3 Calculated 280 Volatile Organic Compound (VOC) g/l EU / EPA 805 Flash Point (Abel) °C IP 170 -6 Auto Ignition Temperature °C ASTM E659 515