Interleukin-17 Receptor a Signaling in Transformed Enterocytes Promotes Early Colorectal Tumorigenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Interleukin 22 Pathway Interacts with Mutant KRAS to Promote Poor Prognosis in Colon Cancer
Author Manuscript Published OnlineFirst on May 19, 2020; DOI: 10.1158/1078-0432.CCR-19-1086 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. The interleukin 22 pathway interacts with mutant KRAS to promote poor prognosis in colon cancer Authors: Sarah McCuaig1, David Barras,2, Elizabeth Mann1, Matthias Friedrich1, Samuel Bullers1, Alina Janney1, Lucy C. Garner1, Enric Domingo3, Viktor Hendrik Koelzer3,4,5, Mauro Delorenzi2,6,7, Sabine Tejpar8, Timothy Maughan9, Nathaniel R. West1, Fiona Powrie1 Affiliations: 1 Kennedy Institute of Rheumatology, University of Oxford, Oxford UK. 2 SIB Swiss Institute of Bioinformatics, Bioinformatics Core Facility, Lausanne, Switzerland. 3Department of Oncology, University of Oxford, Oxford UK. 4Nuffield Department of Medicine, University of Oxford, Oxford UK. 5Department of Pathology and Molecular Pathology, University and University Hospital Zurich, Zurich Switzerland. 6 Ludwig Center for Cancer Research, University of Lausanne, Lausanne, Switzerland. 7 Department of Oncology, Faculty of Biology and Medicine, University of Lausanne, Lausanne Switzerland. 8 Molecular Digestive Oncology, KU Leuven, Belgium. 9 CRUK/MRC Oxford Institute for Radiation Oncology, University of Oxford, Oxford, UK. Downloaded from clincancerres.aacrjournals.org on September 26, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on May 19, 2020; DOI: 10.1158/1078-0432.CCR-19-1086 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Correspondence to: Professor Fiona Powrie; Kennedy Institute of Rheumatology, University of Oxford, Roosevelt Drive, Headington, Oxford, OX3 7YF, UK. Email: [email protected] Conflicts of Interest: S.M., N.R.W., and F.P. -

IL-1Β Induces the Rapid Secretion of the Antimicrobial Protein IL-26 From
Published June 24, 2019, doi:10.4049/jimmunol.1900318 The Journal of Immunology IL-1b Induces the Rapid Secretion of the Antimicrobial Protein IL-26 from Th17 Cells David I. Weiss,*,† Feiyang Ma,†,‡ Alexander A. Merleev,x Emanual Maverakis,x Michel Gilliet,{ Samuel J. Balin,* Bryan D. Bryson,‖ Maria Teresa Ochoa,# Matteo Pellegrini,*,‡ Barry R. Bloom,** and Robert L. Modlin*,†† Th17 cells play a critical role in the adaptive immune response against extracellular bacteria, and the possible mechanisms by which they can protect against infection are of particular interest. In this study, we describe, to our knowledge, a novel IL-1b dependent pathway for secretion of the antimicrobial peptide IL-26 from human Th17 cells that is independent of and more rapid than classical TCR activation. We find that IL-26 is secreted 3 hours after treating PBMCs with Mycobacterium leprae as compared with 48 hours for IFN-g and IL-17A. IL-1b was required for microbial ligand induction of IL-26 and was sufficient to stimulate IL-26 release from Th17 cells. Only IL-1RI+ Th17 cells responded to IL-1b, inducing an NF-kB–regulated transcriptome. Finally, supernatants from IL-1b–treated memory T cells killed Escherichia coli in an IL-26–dependent manner. These results identify a mechanism by which human IL-1RI+ “antimicrobial Th17 cells” can be rapidly activated by IL-1b as part of the innate immune response to produce IL-26 to kill extracellular bacteria. The Journal of Immunology, 2019, 203: 000–000. cells are crucial for effective host defense against a wide and neutrophils. -

Proinflammatory T Helper Type 17 Cells Are Effective B-Cell Helpers
Proinflammatory T helper type 17 cells are effective B-cell helpers Meike Mitsdoerffera, Youjin Leea, Anneli Jägera, Hye-Jung Kimb, Thomas Kornc, Jay K. Kollsd, Harvey Cantorb,1, Estelle Bettellie,1, and Vijay K. Kuchrooa,1 aCenter for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; bDana-Farber Cancer Institute, Boston, MA 02115; cDepartment of Neurology, Technical University Munich, 81675 Munich, Germany; dLouisiana State University Health Sciences Center, New Orleans, LA 70112; and eBenaroya Research Institute, Seattle, WA 98101 Contributed by Harvey Cantor, July 1, 2010 (sent for review May 11, 2010) T helper type 17 (TH17) cells are highly proinflammatory effector collaboration. TH1 and TH2 cells are known to provide B-cell T cells that are characterized by the production of high amounts of help, and their signature cytokines IFN-γ and IL-4 induce class IL-17A, IL-17F, IL-21, and IL-22. Furthermore, TH17 cells have been switch recombination to IgG2a or IgG1 and IgE, respectively (9, associated with a number of autoimmune diseases. However, it is 10). Using IFN-γ and IL-4 reporter mice, Reinhardt et al. (11) not clear whether TH17 cells can also serve as effective helper cells. demonstrated that B cells form conjugates with antigen-activated, Here we show that TH17 cells can function as B-cell helpers in cytokine-producing effector T cells and that the profile of cyto- that they not only induce a strong proliferative response of B cells kine production defines isotype class switching in conjugated B in vitro but also trigger antibody production with class switch cells. -

IL-17-Producing Cells in Tumor Immunity: Friends Or Foes?
Immune Netw. 2020 Feb;20(1):e6 https://doi.org/10.4110/in.2020.20.e6 pISSN 1598-2629·eISSN 2092-6685 Review Article IL-17-Producing Cells in Tumor Immunity: Friends or Foes? Da-Sol Kuen 1,2, Byung-Seok Kim 1, Yeonseok Chung 1,2,* 1Laboratory of Immune Regulation, Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Korea 2BK21 Plus Program, Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Korea Received: Dec 29, 2019 Revised: Jan 25, 2020 ABSTRACT Accepted: Jan 26, 2020 IL-17 is produced by RAR-related orphan receptor gamma t (RORγt)-expressing cells *Correspondence to including Th17 cells, subsets of γδT cells and innate lymphoid cells (ILCs). The biological Yeonseok Chung significance of IL-17-producing cells is well-studied in contexts of inflammation, Laboratory of Immune Regulation and BK21 Plus Program, Institute of Pharmaceutical autoimmunity and host defense against infection. While most of available studies in tumor + Sciences, Seoul National University, 1 Gwanak- immunity mainly focused on the role of T-bet-expressing cells, including cytotoxic CD8 ro, Gwanak-gu, Seoul 08826, Korea. T cells and NK cells, and their exhaustion status, the role of IL-17-producing cells remains E-mail: [email protected] poorly understood. While IL-17-producing T-cells were shown to be anti-tumorigenic in Copyright © 2020. The Korean Association of adoptive T-cell therapy settings, mice deficient in type 17 genes suggest a protumorigenic Immunologists potential of IL-17-producing cells. This review discusses the features of IL-17-producing This is an Open Access article distributed cells, of both lymphocytic and myeloid origins, as well as their suggested pro- and/or anti- under the terms of the Creative Commons tumorigenic functions in an organ-dependent context. -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -

IL-25-Induced Activities IL-17RB and IL-17RA in Mediating Identification of Functional Roles for Both
Identification of Functional Roles for Both IL-17RB and IL-17RA in Mediating IL-25-Induced Activities This information is current as Erika A. Rickel, Lori A. Siegel, Bo-Rin Park Yoon, James B. of September 29, 2021. Rottman, David G. Kugler, David A. Swart, Penny M. Anders, Joel E. Tocker, Michael R. Comeau and Alison L. Budelsky J Immunol 2008; 181:4299-4310; ; doi: 10.4049/jimmunol.181.6.4299 Downloaded from http://www.jimmunol.org/content/181/6/4299 References This article cites 30 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/181/6/4299.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 29, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Identification of Functional Roles for Both IL-17RB and IL-17RA in Mediating IL-25-Induced Activities Erika A. -

The Interleukin 22 Pathway Interacts with Mutant KRAS to Promote Poor Prognosis in Colon Cancer
Author Manuscript Published OnlineFirst on May 19, 2020; DOI: 10.1158/1078-0432.CCR-19-1086 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. The interleukin 22 pathway interacts with mutant KRAS to promote poor prognosis in colon cancer Authors: Sarah McCuaig1, David Barras,2, Elizabeth Mann1, Matthias Friedrich1, Samuel Bullers1, Alina Janney1, Lucy C. Garner1, Enric Domingo3, Viktor Hendrik Koelzer3,4,5, Mauro Delorenzi2,6,7, Sabine Tejpar8, Timothy Maughan9, Nathaniel R. West1, Fiona Powrie1 Affiliations: 1 Kennedy Institute of Rheumatology, University of Oxford, Oxford UK. 2 SIB Swiss Institute of Bioinformatics, Bioinformatics Core Facility, Lausanne, Switzerland. 3Department of Oncology, University of Oxford, Oxford UK. 4Nuffield Department of Medicine, University of Oxford, Oxford UK. 5Department of Pathology and Molecular Pathology, University and University Hospital Zurich, Zurich Switzerland. 6 Ludwig Center for Cancer Research, University of Lausanne, Lausanne, Switzerland. 7 Department of Oncology, Faculty of Biology and Medicine, University of Lausanne, Lausanne Switzerland. 8 Molecular Digestive Oncology, KU Leuven, Belgium. 9 CRUK/MRC Oxford Institute for Radiation Oncology, University of Oxford, Oxford, UK. Downloaded from clincancerres.aacrjournals.org on September 30, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on May 19, 2020; DOI: 10.1158/1078-0432.CCR-19-1086 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Correspondence to: Professor Fiona Powrie; Kennedy Institute of Rheumatology, University of Oxford, Roosevelt Drive, Headington, Oxford, OX3 7YF, UK. Email: [email protected] Conflicts of Interest: S.M., N.R.W., and F.P. -

ABD-Derived Protein Blockers of Human IL-17 Receptor a As Non-Igg Alternatives for Modulation of IL-17-Dependent Pro-Inflammatory Axis
International Journal of Molecular Sciences Article ABD-Derived Protein Blockers of Human IL-17 Receptor A as Non-IgG Alternatives for Modulation of IL-17-Dependent Pro-Inflammatory Axis Marie Hlavniˇcková 1, Milan Kuchaˇr 1, Radim Osiˇcka 2, Lucie Vaˇnková 1, Hana Petroková 1, Michal Malý 1, Jiˇrí Cernˇ ý 3 , Petr Arenberger 4 and Petr Malý 1,* 1 Laboratory of Ligand Engineering, Institute of Biotechnology of the Czech Academy of Sciences, v. v. i., BIOCEV Research Center, Pru˚myslová 595, 252 50 Vestec, Czech Republic; [email protected] (M.H.); [email protected] (M.K.); [email protected] (L.V.); [email protected] (H.P.); [email protected] (M.M.) 2 Laboratory of Molecular Biology of the Bacterial Pathogens, Institute of Microbiology, Czech Academy of Sciences, v. v. i., Vídeˇnská 1083, 142 20 Prague, Czech Republic; [email protected] 3 Laboratory of Structural Bioinformatics of Proteins, Institute of Biotechnology of the Czech Academy of Sciences, v. v. i., BIOCEV Research Center, Pru˚myslová 595, 252 50 Vestec, Czech Republic; [email protected] 4 Department of Dermatology and Venereology, Faculty Hospital of Královské Vinohrady, Šrobárova 50, 100 34 Prague, Czech Republic; [email protected] * Correspondence: [email protected]; Tel.: +420-325-873-763 Received: 10 September 2018; Accepted: 6 October 2018; Published: 9 October 2018 Abstract: Interleukin 17 (IL-17) and its cognate receptor A (IL-17RA) play a crucial role in Th17 cells-mediated pro-inflammatory pathway and pathogenesis of several autoimmune disorders including psoriasis. -

IL-17RA-Signaling Modulates CD8+ T Cell Survival and Exhaustion During 2 Trypanosoma Cruzi Infection
bioRxiv preprint doi: https://doi.org/10.1101/314336; this version posted May 11, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 IL-17RA-signaling modulates CD8+ T cell survival and exhaustion during 2 Trypanosoma cruzi infection 3 Jimena Tosello Boari1,2, Cintia L. Araujo Furlan1,2, Facundo Fiocca Vernengo1,2, Constanza 4 Rodriguez1,2, María C. Ramello1,2, María C. Amezcua Vesely1,2, Melisa Gorosito Serrán1,2, 5 Nicolás G. Nuñez3,4, Wilfrid Richer3,4, Eliane Piaggio3,4, Carolina L. Montes1,2, Adriana 6 Gruppi1,2, Eva V. Acosta Rodríguez1,2*. 7 1 Departamento de Bioquímica Clínica. Facultad de Ciencias Químicas, Universidad 8 Nacional de Córdoba, Córdoba, X5000HUA. Argentina. 9 2 Centro de Investigaciones en Bioquímica Clínica e Inmunología. CONICET. Córdoba, 10 X5000HUA. Argentina. 11 3 SiRIC TransImm «Translational Immunotherapy Team», Translational Research 12 Department, Research Center, PSL Research University, INSERM U932, Institut Curie, 13 Paris, 75005. France. 14 4 Centre d’Investigation Clinique Biothérapie CICBT 1428, Institut Curie, Paris, 75005. 15 France. 16 Running title: IL-17RA signaling regulates CD8+ T cell responses to T. cruzi 17 *Corresponding autor: 18 Eva V. Acosta Rodríguez 19 [email protected] 20 1 bioRxiv preprint doi: https://doi.org/10.1101/314336; this version posted May 11, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Il-17 Receptor Composition 1995)
RESEARCH HIGHLIGHTS Nature Reviews Immunology | Published online 23 November 2015; doi:10.1038/nri.2015.2 shown to signal through IL-17RA, of the IL-17 family, whereas IL-17RC Journal club which was also unrelated to other seems to be specific for IL-17 and known cytokine receptors (Yao et al., IL-17F. Nonetheless, there are few IL-17 RECEPTOR COMPOSITION 1995). However, it was unclear how phenotypic differences between mice IL-17RA operated at a molecular level. lacking IL-17RA or IL-17RC, and It has now been a decade since the Toy et al. approached this issue by patients with null mutations in either paradigm-shifting discovery of T helper reconstituting fibroblasts from Il17ra–/– subunit also have remarkably similar 17 (TH17) cells in 2005 forced everyone mice with human IL-17RA. They defects, with their major phenotype working on effector CD4+ T cells to observed that IL-17-induced signalling being chronic mucosal candidiasis reinterpret their findings in the context could only be restored in these (Puel et al., 2011; Ling et al., 2015). of this interleukin-17 (IL-17)-producing fibroblasts upon co-expression of Thus, this important study by Toy et al. TH cell population. We now know that human IL-17RC with human IL-17RA, clarified the constituents of the IL-17 IL-17-induced signalling has a crucial but not upon co-expression of other receptor complex, which set the stage role in immunity to extracellular IL-17R family members. As the Il17ra–/– for a deeper understanding of its microorganisms, particularly to fungal cells expressed endogenous mouse biological activities. -
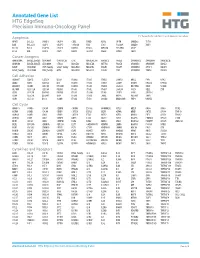
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

Interleukin-17C Promotes Th17 Cell Responses and Autoimmune Disease Via Interleukin-17 Receptor E
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Immunity Article Interleukin-17C Promotes Th17 Cell Responses and Autoimmune Disease via Interleukin-17 Receptor E Seon Hee Chang,1,2 Joseph M. Reynolds,1,2 Bhanu P. Pappu,1 Guangjie Chen,1 Gustavo J. Martinez,1 and Chen Dong1,* 1Department of Immunology and Center for Inflammation and Cancer, MD Anderson Cancer Center, Houston, TX 77054-1901, USA 2These authors contributed equally to this work *Correspondence: [email protected] DOI 10.1016/j.immuni.2011.09.010 SUMMARY IL-23 (Langrish et al., 2005) for their differentiation and cytokine expression. Although several interleukin-17 (IL-17) family Th17 cells, via production of IL-17A and IL-17F, promote the members and their receptors have been recently development of autoimmune diseases while protecting the appreciated as important regulators in inflammatory host against bacterial and fungal infections (Kolls and Linde´ n, diseases, the function of other IL-17 cytokines and 2004). IL-17A and IL-17F signal through the IL-17 receptor A IL-17 receptor-like molecules is unclear. Here we (IL-17RA)-IL-17RC complex (Hu et al., 2010; Toy et al., 2006). show that an IL-17 cytokine family member, IL-17C, Although IL-17RA is expressed ubiquitously, IL-17RC expres- sion is dominant in nonhematopoietic cells (Kuestner et al., was induced in a Th17 cell-dependent autoimmune 2007). IL-17A and IL-17F target epithelial cells or fibroblasts to disease and was required for its pathogenesis. produce arrays of cytokines and chemokines including IL-6 IL-17C bound to IL-17RE, a member of IL-17 receptor and CXCL1 (Chang and Dong, 2009; Gaffen, 2008).