A General Stereoselective Approach to 1,2,4-Triazepane-3-Thiones/Ones
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent 0
1 3,667,923 United States Patent 0 ice Patented June 6, 1972 1 2 ‘I have discovered that anhydrous hydrogen cyanide $667,923 reacts readily with a quaternary ammonium borohydride PREPARATION OF LITHIUM, SODIUM AND and with the borohydrides of lithium and sodium at QUATERNARY AMMONIUM CYANOBORO atmospheric pressure in solvents for the borohydride, such HYDRIDES Robert C. Wade, Ipswich, Mass, assignor to Ventron as glyme, diglyme, triglyme, tetrahydrofuran and dimethyl Corporation, Beverly, Mass. formamide, or mixtures of these at a temperatures be No Drawing. Filed June 16, 1969, Ser. No. 833,722 tween 0° and the boiling point of the solvent to form Int. Cl. C01c 3/08; C01!) 35/00 the corresponding cyanoborohydride. The preferred sol US. Cl. 23-358 4 Claims vents are tetrahydrofuran and glyme because of their 10 convenient boiling points. Potassium borohydride does not react readily with ABSTRACT OF THE DISCLOSURE hydrogen cyanide in tetrahydrofuran or glyme presum The invention relates to the preparation of lithium, ably because of its lack of solubility in these solvents. sodium and quaternary ammonium cyanoborohydrides. However, it reacts with hydrogen cyanide in dimethyl These compounds are prepared by mixing substantially formamide similarly to the other borohydrides mentioned anhydrous hydrogen cyanide with a substantially an above. hydrous lithium or sodium or quaternary ammonium The reaction of the process of the invention appears to borohydride at a temperature between 0° C. and 100° C. take place in two stages. Thus, if the reaction mixture is in a substantially anhydrous solvent, such as tetrahydro initially maintained between about 10° and about 35° C. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Part I. the Total Synthesis Of
AN ABSTRACT OF THE THESIS OF Lester Percy Joseph Burton forthe degree of Doctor of Philosophy in Chemistry presentedon March 20, 1981. Title: Part 1 - The Total Synthesis of (±)-Cinnamodialand Related Drimane Sesquiterpenes Part 2 - Photochemical Activation ofthe Carboxyl Group Via NAcy1-2-thionothiazolidines Abstract approved: Redacted for privacy DT. James D. White Part I A total synthesis of the insect antifeedant(±)-cinnamodial ( ) and of the related drimanesesquiterpenes (±)-isodrimenin (67) and (±)-fragrolide (72)are described from the diene diester 49. Hydro- boration of 49 provided the C-6oxygenation and the trans ring junction in the form of alcohol 61. To confirm the stereoselectivity of the hydroboration, 61 was convertedto both (t)-isodrimenin (67) and (±)-fragrolide (72) in 3 steps. A diisobutylaluminum hydride reduction of 61 followed by a pyridiniumchlorochromate oxidation and treatment with lead tetraacetate yielded the dihydrodiacetoxyfuran102. The base induced elimination of acetic acid preceded theepoxidation and provided 106 which contains the desired hydroxy dialdehydefunctionality of cinnamodial in a protected form. The epoxide 106 was opened with methanol to yield the acetal 112. Reduction, hydrolysis and acetylation of 112 yielded (t)- cinnamodial in 19% overall yield. Part II - Various N- acyl- 2- thionothiazolidineswere prepared and photo- lysed in the presence of ethanol to provide the corresponding ethyl esters. The photochemical activation of the carboxyl function via these derivatives appears, for practical purposes, to be restricted tocases where a-keto hydrogen abstraction and subsequent ketene formation is favored by acyl substitution. Part 1 The Total Synthesis of (±)-Cinnamodial and Related Drimane Sesquiterpenes. Part 2 Photochemical Activation of the Carboxyl Group via N-Acy1-2-thionothiazolidines. -
![Reductive Amination with [11C]Formaldehyde: a Versatile Approach to Radiomethylation of Amines](https://docslib.b-cdn.net/cover/4140/reductive-amination-with-11c-formaldehyde-a-versatile-approach-to-radiomethylation-of-amines-684140.webp)
Reductive Amination with [11C]Formaldehyde: a Versatile Approach to Radiomethylation of Amines
International Journal of Organic Chemistry, 2012, 2, 202-223 http://dx.doi.org/10.4236/ijoc.2012.23030 Published Online September 2012 (http://www.SciRP.org/journal/ijoc) Reductive Amination with [11C]Formaldehyde: A Versatile Approach to Radiomethylation of Amines Chunying Wu1, Ruoshi Li1, Dorr Dearborn2, Yanming Wang1* 1Division of Radiopharmaceutical Science, Case Center for Imaging Research, Department of Radiology, Cleveland, USA 2Environmental Health, Case Western Reserve University, Cleveland, USA Email: *[email protected] Received February 16, 2012; revised March 24, 2012; accepted April 2, 2012 ABSTRACT Carbon-11 radiolabeled amines constitute a very important class of radioligands that are widely used for positron emis- 11 sion tomography (PET) imaging. Radiolabeling of amines is often achieved through radiomethylation using [ C]CH3I 11 or [ C]CH3OTf under basic conditions in a strictly anhydrous environment. Functional groups such as hydroxyl and carboxyl groups that are often present in the molecules are normally base sensitive and require protection and deprotec- tion, which substantially prolongs and complicates the radiolabeling process. Here we report a versatile approach to a series of C-11 radiolabeled amines prepared through reductive amination using [11C]formaldehyde. Using a variety of substrates bearing different functional groups, we demonstrate the general utility of this method. In contrast to conven- tional radiomethylation methods, the reductive amination using [11C]formaldehyde can be carried out in an aqueous environment relatively quickly without the need of protection of base-sensitive functional groups. Keywords: C-11 Formaldehyde; Radiomethylation; Reductive Amination; Positron Emission Tomography; Radiolabelling 1. Introduction probes must be labeled with positron-emitting radionu- clides such as carbon-11 or fluorine-18. -

Safe Handling and Disposal of Chemicals Used in the Illicit Manufacture of Drugs
Vienna International Centre, PO Box 500, 1400 Vienna, Austria Tel.: (+43-1) 26060-0, Fax: (+43-1) 26060-5866, www.unodc.org Guidelines for the Safe handling and disposal of chemicals used in the illicit manufacture of drugs United Nations publication USD 26 Printed in Austria ISBN 978-92-1-148266-9 Sales No. E.11.XI.14 ST/NAR/36/Rev.1 V.11-83777—September*1183777* 2011—300 Guidelines for the Safe handling and disposal of chemicals used in the illlicit manufacture of drugs UNITED NATIONS New York, 2011 Symbols of United Nations documents are composed of letters combined with figures. Mention of such symbols indicates a reference to a United Nations document. ST/NAR/36/Rev.1 UNITED NATIONS PUBLICATION Sales No. E.11.XI.14 ISBN 978-92-1-148266-9 eISBN 978-92-1-055160-1 © United Nations, September 2011. All rights reserved. The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. Requests for permission to reproduce this work are welcomed and should be sent to the Secretary of the Publications Board, United Nations Headquarters, New York, N.Y. 10017, U.S.A. or also see the website of the Board: https://unp.un.org/Rights.aspx. Governments and their institutions may reproduce this work without prior authoriza- tion but are requested to mention the source and inform the United Nations of such reproduction. -

Analogues of Steffimycinone
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 4-1982 Analogues of Steffimycinone David Wayne Elrod Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Pharmacology Commons Recommended Citation Elrod, David Wayne, "Analogues of Steffimycinone" (1982). Master's Theses. 1697. https://scholarworks.wmich.edu/masters_theses/1697 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. ANALOGUES OF STEFFIMYCINONE by David Wayne Elrod A Thesis Submitted to the Faculty of The Graduate College in partial fulfillment of the requirements for the Degree of Master of Arts Department o f Chemistry Western Michigan U niversity Kalamazoo, Michigan A pril 1982 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ANALOGUES OF STEFFIMYCINONE David Wayne Elrod, M.A. Western Michigan U niversity, 1982 Several analogues o f Steffimycinone - an anthracycline antitumor antibiotic - have been prepared. Modifications were made at C-7 and C-10 of the non-aromatic A ring portion of the anthracycline nucleus. Compounds with nitrogen substituents were prepared. The synthesis of these compounds is discussed as well as their antitumor activity. The unsuccessful approaches to several other analogues are also discussed. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGMENTS I would lik e to express my appreciation to Dr. Paul F. Wiley of The Upjohn Company fo r his in sp ira tio n , guidance and encouragement. -

Aminolink™Reductant
INSTRUCTIONS AminoLink ™ Reductant (s odium cyanoborohydride, NaCNBH ) 3 1291.3 44892 Number Description 44892 AminoLink Reductant (sodium cyanoborohydride, NaCNBH3), 2 × 1g Form: white to yellow crystalline powder; may yield slightly hazy solution in water Molecular Weight: 62.84 CAS # 25895-60-7 Caution: NaCNBH3 is toxic and must be used in a fume hood. See the Material Safety Data Sheet (MSDS) for additional information. Storage: Upon receipt store desiccated at room temperature. Product shipped at ambient temperature. Introduction Thermo Scientific AminoLink Reductant promotes the formation of stable bonds between aldehyde- and amine-containing molecules, enabling efficient labeling, conjugation and immobilization of proteins and other research molecules. Carbonyl groups such as aldehydes, ketones and glyoxals react spontaneously with amino groups to form Schiff base intermediates that are in equilibrium with their free forms. The interaction is pH-dependent, being somewhat more efficient in acidic conditions and especially strong at high pH. Addition of AminoLink Reductant (sodium cyanoborohydride) to a reaction in which Schiff base formation has occurred results in complete reduction of the labile Schiff base intermediate to a chemically stable bond1 (Figure 1). Unlike sodium borohydride, sodium cyanoborohydride is sufficiently mild to avoid adversely reducing aldehydes to nonreactive hydroxyls. The overall reaction of carbonyl groups to primary amino groups using sodium cyanoborohydride is called reductive amination. Immobilization by reductive amination of amine-containing biological molecules onto aldehyde-containing solid supports is used to create matrices for affinity purification of antibodies and other molecules. For detailed immobilization procedures, consult the instructions for AminoLink Plus Coupling Resin or the AminoLink Plus Immobilization Kit (see Related Thermo Scientific Products). -

Sodium Cyanoborohydride and Sodium Borohydride
FT-05777C Sodium CyanoBoroHydride and Sodium BoroHydride Products Information Sodium CyanoBoroHydride and Sodium BoroHydride are popular reducing agents for a variety applications Catalog #: 05777C, 5g Name: Sodium cyanoborohydride. CAS:25895-60-7 Molecular Weight : MW : 62.84 Solubility Soluble in water (100 mg/ml, with heating), methanol, ethanol, and THF. It is insoluble in nonpolar solvents such as benzene or hexane other names: Storage: -20°C (or 4°C short term). (M) cyanotrihydroborate Protect from moisture. Keep dry UN:3134, II, 4.3, 6.1 Catalog #: 07998H, 5 g Name: Sodium BoroHydride (NaBH4) . CAS:16940-66-2 / 15681-89-7 Molecular Weight : MW : 37.83 Solubility Soluble in water (100 mg/ml, with heating), methanol, ethanol, and THF. It is insoluble in nonpolar solvents such as benzene or hexane Storage: -20°C (or 4°C short term). (M) other names: Protect from moisture. Keep dry cyanotetrahydroborate UN:3134, II, 4.3, 6.1 Catalog #: 22818F Name: Sodium TriAcetOxyBorohydride CAS: 56553-60-7; Molecular Weight : MW: 211.94 Technical and Scientific Information ●Sodium CyanoBoroHydride Sodium CyanoBoroHydride (NaBH3CN) is a selective reducing agent used for a variety of chemical reductions, including aldehyde, ketones, oximes, enamines, reductive aminations of aldehydes and ketones, and reductive alkylations of amines and hydrazines. The utility of sodium cyanoborohydride as a reducing agent is greatly enhanced by its stability under acid conditions, and its solubility in aprotic solvents. Sodium cyanoborohydride is a milder and more selective reducing agent than sodium borohydride () 1. Reduction of aldehydes and ketones. At pH 3-4, benzaldehyde can be reduced to benzyl alcohol with 87% yield. -

Kemira Sodium Borohydride Nabh4 an Effective Reducing Agent Sodium Borohydride Nabh4
Kemira Sodium Borohydride NaBH4 An effective reducing agent Sodium Borohydride NaBH4 An effective and selective reducing agent Reduction of organic compounds NaBH4 is a very effective and selective reducing agent. It is In organic reactions the reduction occurs on the carbon atom widely used in its dry form (white crystalline powder or granules) having the highest positive partial charge. The rate of reduction by the pharmaceutical and fine chemical industry in applications is increased by any substituent that increases the partial posi- where an excellent hydrogenation and reduction is needed. tive charge of the carbonyl carbon. The reductions of carbonyl The reductions can be carried out with a wide range of carbo- groups by borohydride occur mostly by nucleophilic attack of nyl substrates and the products are often building blocks espe- hydride on the carbonyl carbon. cially for the synthesis of pharmaceutically active compounds. The basic reduction mechanism of sodium borohydride. NaBH4 is also used to purify organic chemicals by removing metal ions, carbonyl and peroxide impurities that cause unde- R R sirable odor, taste, color and instability to products. 4 H2O/ROH - 4 C = O + BH4 4 C OH Sodium borohydride is easy to handle when the appropriate safety precautions are followed. It is not necessary to exclude H moisture nor atmospheric oxygen when using NaBH4. The wa- ter present in the reaction mixture usually does not have unfa- The mechanism shows that 1 mol of NaBH4 can reduce as vourable effects on the desired reduction, provided that there many as four molecules of a carbonyl compound. Four mol- is sufficient borohydride present. -

D:\Firoz\Materials of All Topic
Reductions 37 R SPH Na, Li R H NH3 O S Na, NH3(l) R NH R' R NH2 Na, NH3 R2 N Ts R 2 NH 2.9. Reduction by hyride transfer-reagent: REACTIVITY OF HYDRIDE DONAR REDUCING AGENTS Iminium Acyl Aldehyde Ester Amide Caboxylic ion chloride or ketone salt Hydride Donar More Reactive Least Reactive LiAlH4 Amine Alcohol Alcohol Alcohol Amine Alcohol Red Al Alcohol Alcohol Alcohol Amine Alcohol LiAlH (Ot-Bu4)3 Aldehyde Alcohol Alcohol Aldehyde NaBH4 Amine Alcohol Alcohol NaBH3CN Amine B2H6 Alcohol Amine Alcohol AlH3 Alcohol Alcohol Alcohol Amine Alcohol Disiamylborane Alcohol Aldehyde DIBAL Alcohol Aldehyde Aldehyde Alcohol 2.9.1. Lithium aluminium hydride (LiAlH4) Most reductions of carbonyl compounds are done with reagents that transfer a hydride from boron or aluminium. Sodium borohydride is a mild reducing reagent that rapidly reduce aldehyde and ketones but not esters. Lithium aluminium is strongly donor reagent and it rapidly reduce ester acids, nitriles, amides as well as aldehyde and ketones. Neither sodium borohydride nor lithium aluminium hydride reacts with isolated carbon- carbon double bonds. There reagents are nucleophilic and as such they normally attack polarized multiple bond such as C=O, CN by transfer of hydride ion to the more positive atom. Lithium aluminium hydride is a more powerful reducing agent than sodium borohydride and reduces most of commonly encountered organic functional group. O O O AlH3 O O O H H Al 2 3 AlH2 AlH H H O + OH H3O O 4 Al 38 Reductions Common Functional Groups, reduced by LAH Functional groups Reduction product RCHO R CH2OH R R C O CH OH R R O R CH2OH + R'OH R C OR' O RCH OH R C OH 2 O R CH NHR' R C NHR' 2 O R CH2NR'2 or RCH(OH)NR'2 RCHO + R'2(NH) R C NR'2 R CH2 NH2 or R C NH RCHO R C N H R C N OH H2 H R C NH2 ArNO2 Ar NH NH Ar or Ar N N Ar R CH2 Br R CH3 O R CH O S Ar 2 R CH3 O O OH R C CH 2 C CH3 H R H Aldehye, ketone, esters, carboxylic acids and lactones can all be reduced smoothly to corresponding alcohols under mild conditions. -
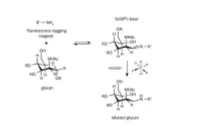
A Novel Carbohydrate Labeling Method Utilizing Transfer Hydrogenation-Mediated Reductive Amination
A NOVEL CARBOHYDRATE LABELING METHOD UTILIZING TRANSFER HYDROGENATION-MEDIATED REDUCTIVE AMINATION Zsuzsanna Kovács1, Gábor Papp2, Henrietta Horváth3, Ferenc Joó2,3 and András Guttman1 1Horváth Csaba Laboratory of Bioseparation Sciences, University of Debrecen, Hungary 2Department of Physical Chemistry, University of Debrecen, Hungary 3MTA-DE Homogeneous Catalysis and Reaction Mechanisms Research Group, University of Debrecen, Hungary Abstract One of the most frequently used high-resolution glycan analysis methods in the biopharmaceutical and biomedical fields is capillary electrophoresis with laser-induced fluorescence (CE-LIF) detection. Glycans are usually labeled by reductive amination with a charged fluorophore containing a primary amine, which reacts with the aldehyde group at the reducing end of the glycan structures. In this reaction, first a Schiff base is formed that is reduced to form a stable conjugate by a hydrogenation reagent, such as sodium cyanoborohydride. In large scale biopharmaceutical applications, such as clone selection for glycoprotein therapeutics, hundreds of reactions are accomplished simultaneously, so the HCN generated in the process poses a safety concern. To alleviate this issue, here we propose catalytic hydrogen transfer from formic acid catalyzed by water-soluble iridium(III)- and ruthenium(II)-phosphine complexes as a novel alternative to hydrogenation. The easily synthesized water-soluble iridium(III) and the ruthenium(II) hydrido complexes showed high catalytic activity in carbohydrate labeling. This procedure is environmentally friendly and reduces the health risks for the industry. Using carbohydrate standards, oligosaccharides released from glycoproteins with highly sialylated (fetuin), high mannose (ribonuclease B) and mixed sialo and neutral (human plasma) N-glycans, we demonstrated similar labeling efficiencies for iridium(III) dihydride to that of the conventionally used sodium cyanoborohydride based reaction. -

Vollhardt Group
Publication List 1. Baxter, C. S.; Garratt, P. J.; Vollhardt, K. P. C., The 3,4:5,6:9,10- Tribenzobicyclo[6.2.0]decapentaenyl Dianion, J. Am. Chem. Soc. 1969, 91, 7783. 2. Garratt, P. J.; Vollhardt, K. P. C., Benzo[3,4]cyclobuta[1,2-c]thiophen (2-Thianorbiphenylene), Chem. Commun. 1970, 109. 3. Garratt, P. J.; Vollhardt, K. P. C.; Mitchell, R. H., Wittig Reactions of 1,2-Dihydro-1,2- bis(triphenylphosphoranylidene)benzocyclobutene and Benzocyclobutene-1,2-quinone. The Synthesis of Dibenzo[a,c]benzo[3,4]cyclobuta[1,2-f]cyclooctene, J. Chem. Soc. (C) 1970, 2137. 4. Garratt, P. J.; Holmes, A. B.; Sondheimer, F.; Vollhardt, K. P. C., The Synthesis of Benzo[3,4]cyclobuta[1,2-e]dicyclohexeno[b,h]thionin, an Analog of Biphenylene Containing a Thionin Ring, J. Am. Chem. Soc. 1970, 92, 4492. 5. Garratt, P. J.; Vollhardt, K. P. C., 6,11-Methanocyclobutabenzo[1,2-a]-[10]annulene, a Biphenylene-Analog Containing a Carbocyclic 10 π-Electron Ring, Angew. Chem. 1971, 83, 111; Angew. Chem., Int. Ed. Engl. 1971, 10, 125. 6. Garratt, P. J.; Vollhardt, K. P. C., Homophthalaldehyde, Synthesis 1971, 3, 423. 7. Garratt, P. J.; Holmes, A. B.; Sondheimer, F.; Vollhardt, K. P. C., The Synthesis of Mono-trans- dicyclohexeno[b,i]-1,6-dithiacyclodeca-2,4,7,9-tetraene, a Tetra-alkylated 1,6-Dithia[10]annulene, Chem. Commun. 1971, 947. 8. Garratt, P. J.; Vollhardt, K. P. C., Synthesis of Benzo[3,4]cyclobuta[1,2-a]benzo[d]tropylium Cation, a Homobiphenylene Cation, Chem.