M5 Keratin 18
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Background Effects of Keratin 8 and 18 in a DDC-Induced Hepatotoxicity and Mallory-Denk Body Formation Mouse Model
Laboratory Investigation (2012) 92, 857–867 & 2012 USCAP, Inc All rights reserved 0023-6837/12 $32.00 Genetic background effects of keratin 8 and 18 in a DDC-induced hepatotoxicity and Mallory-Denk body formation mouse model Johannes Haybaeck1, Cornelia Stumptner1, Andrea Thueringer1, Thomas Kolbe2, Thomas M Magin3, Michael Hesse4, Peter Fickert5, Oleksiy Tsybrovskyy1, Heimo Mu¨ller1, Michael Trauner5,6, Kurt Zatloukal1 and Helmut Denk1 Keratin 8 (K8) and keratin 18 (K18) form the major hepatocyte cytoskeleton. We investigated the impact of genetic loss of either K8 or K18 on liver homeostasis under toxic stress with the hypothesis that K8 and K18 exert different functions. krt8À/À and krt18À/À mice crossed into the same 129-ola genetic background were treated by acute and chronic ad- ministration of 3,5-diethoxy-carbonyl-1,4-dihydrocollidine (DDC). In acutely DDC-intoxicated mice, macrovesicular steatosis was more pronounced in krt8À/À and krt18À/À compared with wild-type (wt) animals. Mallory-Denk bodies (MDBs) appeared in krt18À/À mice already at an early stage of intoxication in contrast to krt8À/À mice that did not display MDB formation when fed with DDC. Keratin-deficient mice displayed significantly lower numbers of apoptotic hepatocytes than wt animals. krt8À/À, krt18À/À and control mice displayed comparable cell proliferation rates. Chronically DDC-intoxicated krt18À/À and wt mice showed a similarly increased degree of steatohepatitis with hepatocyte ballooning and MDB formation. In krt8À/À mice, steatosis was less, ballooning, and MDBs were absent. krt18À/À mice developed MDBs whereas krt8À/À mice on the same genetic background did not, highlighting the significance of different structural properties of keratins. -

'Montalcino, a Zebrafish Model for Variegate Porphyria'
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2008 montalcino, A zebrafish model for variegate porphyria Dooley, Kimberly A ; Fraenkel, Paula G ; Langer, Nathaniel B ; Schmid, Bettina ; Davidson, Alan J ; Weber, Gerhard ; Chiang, Ken ; Foott, Helen ; Dwyer, Caitlin ; Wingert, Rebecca A ; Zhou, Yi ; Paw, Barry H ; Zon, Leonard I Abstract: OBJECTIVE Inherited or acquired mutations in the heme biosynthetic pathway leads to a debilitating class of diseases collectively known as porphyrias, with symptoms that can include anemia, cutaneous photosensitivity, and neurovisceral dysfunction. In a genetic screen for hematopoietic mutants, we isolated a zebrafish mutant, montalcino (mno), which displays hypochromic anemia and porphyria. The objective of this study was to identify the defective gene and characterize the phenotype of the zebrafish mutant. MATERIALS AND METHODS Genetic linkage analysis was utilized to identify the region harboring the mno mutation. Candidate gene analysis together with reverse transcriptase polymerase chain reaction was utilized to identify the genetic mutation, which was confirmed via allele- specific oligo hybridizations. Whole mount in situ hybridizations and o-dianisidine staining were usedto characterize the phenotype of the mno mutant. mRNA and morpholino microinjections were performed to phenocopy and/or rescue the mutant phenotype. RESULTS Homozygous mno mutant embryos have a defect in the protoporphyrinogen oxidase (ppox) gene, which encodes the enzyme that catalyzes the oxidation of protoporphyrinogen. Homozygous mutant embryos are deficient in hemoglobin, and by 36 hours post-fertilization are visibly anemic and porphyric. The hypochromic anemia of mno embryos was partially rescued by human ppox, providing evidence for the conservation of function between human and zebrafish ppox. -
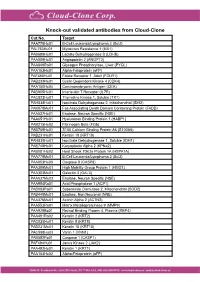
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Motility-Related Actinin Alpha-4 Is Associated with Advanced And
Laboratory Investigation (2008) 88, 602–614 & 2008 USCAP, Inc All rights reserved 0023-6837/08 $30.00 Motility-related actinin alpha-4 is associated with advanced and metastatic ovarian carcinoma Maria V Barbolina1, Brian P Adley2, David L Kelly3, Angela J Fought4, Denise M Scholtens4, Lonnie D Shea1 and M Sharon Stack5 Advanced and metastatic ovarian cancer is a leading cause of death from gynecologic malignancies. A more detailed understanding of the factors controlling invasion and metastasis may lead to novel anti-metastatic therapies. To model cellular interactions that occur during intraperitoneal metastasis, comparative cDNA microarray analysis and confirmatory real-time reverse transcription PCR (RT-PCR) were employed to uncover changes in gene expression that may occur in late stage ovarian cancer in response to microenvironmental cues, particularly native three-dimensional collagen I. Gene expression in human ovarian carcinoma tissues was evaluated on the RNA and protein level using real-time RT-PCR and immunohistochemistry. Cell invasion and migration were evaluated in a collagen invasion assay and a scratch wound assay. Three-dimensional collagen I culture led to differential expression of several genes. The role of actinin alpha-4 (ACTN4), a cytoskeleton-associated protein implicated in the regulation of cell motility, was examined in detail. ACTN4 RNA and protein expression were associated with advanced and metastatic human ovarian carcinoma. This report demonstrates that a cytoskeletal-associated protein ACTN4 is upregulated by three-dimensional collagen culture conditions, leading to increased invasion and motility of ovarian cancer cells. Expression of ACTN4 in human ovarian tumors was found to be associated with advanced-stage disease and peritoneal metastases. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Regulation of Keratin Filament Network Dynamics
Regulation of keratin filament network dynamics Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation vorgelegt von Diplom Biologe Marcin Maciej Moch aus Dzierżoniów (früher Reichenbach, NS), Polen Berichter: Universitätsprofessor Dr. med. Rudolf E. Leube Universitätsprofessor Dr. phil. nat. Gabriele Pradel Tag der mündlichen Prüfung: 19. Juni 2015 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar. This work was performed at the Institute for Molecular and Cellular Anatomy at University Hospital RWTH Aachen by the mentorship of Prof. Dr. med. Rudolf E. Leube. It was exclusively performed by myself, unless otherwise stated in the text. 1. Reviewer: Univ.-Prof. Dr. med. Rudolf E. Leube 2. Reviewer: Univ.-Prof. Dr. phil. nat. Gabriele Pradel Ulm, 15.02.2015 2 Publications Publications Measuring the regulation of keratin filament network dynamics. Moch M, and Herberich G, Aach T, Leube RE, Windoffer R. 2013. Proc Natl Acad Sci U S A. 110:10664-10669. Intermediate filaments and the regulation of focal adhesion. Leube RE, Moch M, Windoffer R. 2015. Current Opinion in Cell Biology. 32:13–20. "Panta rhei": Perpetual cycling of the keratin cytoskeleton. Leube RE, Moch M, Kölsch A, Windoffer R. 2011. Bioarchitecture. 1:39-44. Intracellular motility of intermediate filaments. Leube RE, Moch M, Windoffer R. Under review in: The Cytoskeleton. Editors: Pollard T., Dutcher S., Goldman R. Cold Springer Harbor Laboratory Press, Cold Spring Harbor. Multidimensional monitoring of keratin filaments in cultured cells and in tissues. Schwarz N, and Moch M, Windoffer R, Leube RE. -

Supplementary Material Contents
Supplementary Material Contents Immune modulating proteins identified from exosomal samples.....................................................................2 Figure S1: Overlap between exosomal and soluble proteomes.................................................................................... 4 Bacterial strains:..............................................................................................................................................4 Figure S2: Variability between subjects of effects of exosomes on BL21-lux growth.................................................... 5 Figure S3: Early effects of exosomes on growth of BL21 E. coli .................................................................................... 5 Figure S4: Exosomal Lysis............................................................................................................................................ 6 Figure S5: Effect of pH on exosomal action.................................................................................................................. 7 Figure S6: Effect of exosomes on growth of UPEC (pH = 6.5) suspended in exosome-depleted urine supernatant ....... 8 Effective exosomal concentration....................................................................................................................8 Figure S7: Sample constitution for luminometry experiments..................................................................................... 8 Figure S8: Determining effective concentration ......................................................................................................... -

A Dysfunctional Desmin Mutation in a Patient with Severe Generalized Myopathy
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 11312–11317, September 1998 Genetics A dysfunctional desmin mutation in a patient with severe generalized myopathy ANA M. MUN˜OZ-MA´RMOL*†‡,GERALDINE STRASSER‡§,MARCOS ISAMAT*†,PIERRE A. COULOMBE¶,YANMIN YANG§, i XAVIER ROCA†,ELENA VELA*†,JOSE´ L. MATE†,JAUME COLL ,MARI´A TERESA FERNA´NDEZ-FIGUERAS†, JOSE´ J. NAVAS-PALACIOS†,AURELIO ARIZA†, AND ELAINE FUCHS§** *Fundacio´n Echevarne, 08037 Barcelona, Spain; Departments of †Pathology and iNeurology, Hospital Universitari Germans Trias i Pujol, Universitat Auto´noma de Barcelona, 08916 Badalona, Barcelona, Spain; ¶Department of Biological Chemistry, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205; and §Howard Hughes Medical Institute and Department of Molecular Genetics and Cell Biology, The University of Chicago, Chicago, IL 60637 Contributed by Elaine Fuchs, July 29, 1998 ABSTRACT Mice lacking desmin produce muscle fibers desmin functions in cellular transmission of both active and with Z disks and normal sarcomeric organization. However, passive force (14, 15). the muscles are mechanically fragile and degenerate upon Desminopathies are a heterogeneous group of human myop- repeated contractions. We report here a human patient with athies characterized by abnormal aggregations of desmin in severe generalized myopathy and aberrant intrasarcoplasmic skeletal andyor cardiac muscle fibers (for review, see ref. 16). The accumulation of desmin intermediate filaments. Muscle tissue clumping of desmin in muscle cells of these patients bears a from this patient lacks the wild-type desmin allele and has a striking resemblance to the keratin aggregates that accumulate in desmin gene mutation encoding a 7-aa deletion within the degenerating epithelial cells of various human keratin disorders coiled-coil segment of the protein. -

Biological Functions of Cytokeratin 18 in Cancer
Published OnlineFirst March 27, 2012; DOI: 10.1158/1541-7786.MCR-11-0222 Molecular Cancer Review Research Biological Functions of Cytokeratin 18 in Cancer Yu-Rong Weng1,2, Yun Cui1,2, and Jing-Yuan Fang1,2,3 Abstract The structural proteins cytokeratin 18 (CK18) and its coexpressed complementary partner CK8 are expressed in a variety of adult epithelial organs and may play a role in carcinogenesis. In this study, we focused on the biological functions of CK18, which is thought to modulate intracellular signaling and operates in conjunction with various related proteins. CK18 may affect carcinogenesis through several signaling pathways, including the phosphoinosi- tide 3-kinase (PI3K)/Akt, Wnt, and extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) signaling pathways. CK18 acts as an identical target of Akt in the PI3K/Akt pathway and of ERK1/2 in the ERK MAPK pathway, and regulation of CK18 by Wnt is involved in Akt activation. Finally, we discuss the importance of gaining a more complete understanding of the expression of CK18 during carcinogenesis, and suggest potential clinical applications of that understanding. Mol Cancer Res; 10(4); 1–9. Ó2012 AACR. Introduction epithelial organs, such as the liver, lung, kidney, pancreas, The intermediate filaments consist of a large number of gastrointestinal tract, and mammary gland, and are also nuclear and cytoplasmic proteins that are expressed in a expressed by cancers that arise from these tissues (7). In the tissue- and differentiation-dependent manner. The compo- absence of CK8, the CK18 protein is degraded and keratin fi intermediate filaments are not formed (8). -

A Usual Frameshift and Delayed Termination Codon Mutation in Keratin 5 Causes a Novel Type of Epidermolysis Bullosa Simplex with Migratory Circinate Erythema
ORIGINAL ARTICLE See related Commentary on pages v and vii A Usual Frameshift and Delayed Termination Codon Mutation in Keratin 5 Causes a Novel Type of Epidermolysis Bullosa Simplex with Migratory Circinate Erythema Li-Hong Gu,1 Soo-Chan Kim,Ã1 Yoshiro Ichiki, Junsu Park,Ã Miki Nagai, and Yasuo Kitajima Department of Dermatology, Gifu University School of Medicine, Gifu, Japan, and ÃCutaneous Biology Research Institute, Brain Korea 21 Project for Medical Science,Yonsei University College of Medicine, Seoul, Korea We report here two unrelated families in Japan and Kor- dicted to produce a mutant keratin 5 protein with a fra- ea having patients with a unique type of epidermolysis meshift of its terminal 41 amino acids and 35 amino bullosa simplex and a novel mutation in the keratin acids longer than the wild-type keratin 5 protein due gene KRT5, i.e., a frameshift and delayed stop codon to a delayed termination codon. As the same abnormal inconsistent with any subtype described before. The pa- elongated mutant KRT5 gene was found in the inde- tients showed migratory circinate erythema and multi- pendent families, the predicted abnormal elongated ple vesicles on the circular belt-like areas a¡ected by keratin protein is likely to lead to an atypical clinical erythema. Electron microscopy of skin biopsies showed phenotype that has never been reported, possibly by a reduction in the number of keratin intermediate ¢la- interfering with the functional interaction between kera- ments in the basal cells without tono¢lament clumping. tin and its associated proteins. Key words: Dowling^Meara/ We identi¢ed a novel heterozygous deletion mutation pigmentation/tail domain/V2 domain. -

Keratin Gene Expression in Non-Epithelial Tissues Detection with Polymerase Chain Reaction
American Journal ofPathology, Vol. 142, No. 4, April 1993 Copyright C American Societyfor Investigative Pathology Keratin Gene Expression in Non-Epithelial Tissues Detection with Polymerase Chain Reaction S. Thomas Traweek, Jane Liu, and ceUs, or any of the seven normal bone marrows Hector Battifora examined. Dilutional experiments showedPCR to From the Sylvia Cowan Laboratory ofSurgical Pathology, be highly sensitive in the detection of keratin 19 Division ofPathology, City ofHope National Medical gene expression, capable of registering one Center, Duarte, California MCF-7 ceU in 106 HL-60 ceUs. These studies show that variable levels of keratin 8 and 18 gene ex- pression may be detected by PCR in a wide varlety ofnon-epithelial tissues, supportlingprevious im- Keratinfilament are characteristicaly present in munohistochemical and phylogenetic studies. epithelial ceUs and tumors, but have also been de- However, keratin 19 gene expression appears to tected in many normal and neoplastic non- be more restricted and was not evident in any he- epithelial ceU types using immunohistochemical matopoietic ceUs devoid of contaminating stro- techniques. To investigate the validity of this mal elements. these flndings suggest a role for seemingly aberrant protein expression, we ap- PCR in the detection ofepithelialmicrometastasis plied the highly sensitivepolymerase chain reac- in certain sites, particularly bone marrow. (Am tion (PCR) technique to study keratin gene ex- JPathol 1993, 142:1111-1118) pression in a variety of non-epithelial tissues. Total RNA was extracted from nine samples of leiomyosarcoma, four non-Hodgkin's lymphoma, Intermediate filaments (IF) are the principal compo- seven normal bone marrows, normal lymph node, nents of the cytoskeleton in mammalian cells. -

Identification of Keratins and Analysis of Their Expression in Carp and Goldfish: Comparison with the Zebrafish and Trout Keratin Catalog
Cell Tissue Res DOI 10.1007/s00441-005-0031-1 REGULAR ARTICLE Dana M. García . Hermann Bauer . Thomas Dietz . Thomas Schubert . Jürgen Markl . Michael Schaffeld Identification of keratins and analysis of their expression in carp and goldfish: comparison with the zebrafish and trout keratin catalog Received: 21 March 2005 / Accepted: 23 May 2005 # Springer-Verlag Abstract With more than 50 genes in human, keratins zebrafish, and the cyprinid fishes being more similar to each make up a large gene family, but the evolutionary pres- other than to the salmonid trout. Because of the detected sure leading to their diversity remains largely unclear. Nev- similarity of keratin expression among the cyprinid fishes, ertheless, this diversity offers a means to examine the we propose that, for certain experiments, they are inter- evolutionary relationships among organisms that express changeable. Although the zebrafish distinguishes itself as keratins. Here, we report the analysis of keratins expressed being a developmental and genetic/genomic model organ- in two cyprinid fishes, goldfish and carp, by two-dimen- ism, we have found that the goldfish, in particular, is a more sional polyacrylamide gel electrophoresis, complementary suitable model for both biochemical and histological stud- keratin blot binding assay, and immunoblotting. We further ies of the cytoskeleton, especially since goldfish cytoskel- explore the expression of keratins by immunofluorescence etal preparations seem to be more resistant to degradation microscopy. Comparison is made with the keratin expres- than those from carp or zebrafish. sion and catalogs of zebrafish and rainbow trout. The keratins among these fishes exhibit a similar range of mo- Keywords Keratin .