Regulation of Keratin Filament Network Dynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Desmin Interacts Directly with Mitochondria
International Journal of Molecular Sciences Article Desmin Interacts Directly with Mitochondria Alexander A. Dayal 1, Natalia V. Medvedeva 1, Tatiana M. Nekrasova 1, Sergey D. Duhalin 1, Alexey K. Surin 1,2 and Alexander A. Minin 1,* 1 Institute of Protein Research of Russian Academy of Sciences, Vavilova st., 34, 119334 Moscow, Russia; [email protected] (A.A.D.); [email protected] (N.V.M.); [email protected] (T.M.N.); [email protected] (S.D.D.); [email protected] (A.K.S.) 2 Pushchino Branch, Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Prospekt Nauki 6, Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected] Received: 14 October 2020; Accepted: 26 October 2020; Published: 30 October 2020 Abstract: Desmin intermediate filaments (IFs) play an important role in maintaining the structural and functional integrity of muscle cells. They connect contractile myofibrils to plasma membrane, nuclei, and mitochondria. Disturbance of their network due to desmin mutations or deficiency leads to an infringement of myofibril organization and to a deterioration of mitochondrial distribution, morphology, and functions. The nature of the interaction of desmin IFs with mitochondria is not clear. To elucidate the possibility that desmin can directly bind to mitochondria, we have undertaken the study of their interaction in vitro. Using desmin mutant Des(Y122L) that forms unit-length filaments (ULFs) but is incapable of forming long filaments and, therefore, could be effectively separated from mitochondria by centrifugation through sucrose gradient, we probed the interaction of recombinant human desmin with mitochondria isolated from rat liver. Our data show that desmin can directly bind to mitochondria, and this binding depends on its N-terminal domain. -

PSPC1 Potentiates IGF1R Expression to Augment Cell Adhesion and Motility
1 Supplementary information 2 PSPC1 potentiates IGF1R expression to augment cell 3 adhesion and motility 4 Hsin-Wei Jen1,2 , De-Leung Gu 2, Yaw-Dong Lang 2 and Yuh-Shan Jou 1,2,* 5 1 Graduate Institute of Life Sciences, National Defense Medical Center, Taipei, Taiwan 6 2 Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan 7 * Author to whom correspondence should be addressed 8 Cells 2020, 9, x; doi: FOR PEER REVIEW www.mdpi.com/journal/cells Cells 2020, 9, x FOR PEER REVIEW 2 of 10 9 10 11 Supplementary Figure S1: Expression of IGF1R and integrin in PSPC1-expressing or PSPC1-depleted 12 HCC cells by Western blotting analysis 13 (A) Detection of IGF1R protein levels in three PSPC1-knockdown cells Huh7, HepG2 and Mahlavu. (B) 14 Detection of selected integrin expression in PSPC1-overexpressing or PSPC1-depleted HCC cells by using 15 their total cell lysates immunoblotted with specific integrin antibodies as shown. 16 17 18 Supplementary Figure S2: PSPC1-modulated IGF1R downstream signaling in HCC cells. Cells 2020, 9, x FOR PEER REVIEW 3 of 10 19 (A, B) Immunoblotting of IGF1R expression in PSPC1-overexpressing SK-Hep1 and PLC5 cells 20 treated with IGF1R shRNAs. (C, D) Cell migration and adhesion were measured in PSPC1- 21 knockdown Hep3B cells rescued with exogenous expression of IGF1R. Exogenous expression of 22 IGF1R in PSPC1-knockdown Hep3B cells were then applied for detection of altered AKT/ERK 23 signaling including (E) total PSPC1, IGF1R, AKT, ERK, p-IGF1R, p-AKT(S473), and 24 p-ERK(T202/Y204) as well as altered FAK/Src signaling including (F) total FAK, Src, p-FAK(Y397) 25 and p-Src(Y416) by immunoblotting assay. -

Renal Ischemia/Reperfusion Injury: Functional Tissue Preservation by Anti-Activated 1 Integrin Therapy
Renal Ischemia/Reperfusion Injury: Functional Tissue Preservation by Anti-Activated 1 Integrin Therapy Ana Molina,* Marı´a Ubeda,* Marı´a M. Escribese,* Laura Garcı´a-Bermejo,* David Sancho,† ʈ Guillermo Pe´rez de Lema,‡ Fernando Lian˜o,§ Carlos Caban˜as, Francisco Sa´nchez-Madrid,† and Francisco Mampaso* *Department of Pathology, Hospital Ramo´n y Cajal, Universidad de Alcala´, Madrid, Spain; †Department of Immunology, Hospital de la Princesa, Universidad Auto´noma de Madrid, Madrid, Spain; ‡Medizinische Poliklinic der Ludwig Maximillians-Universitat, Munich, Germany; §Department of Nephrology, Hospital Ramo´n y Cajal, ʈ Universidad de Alcala´, Madrid, Spain; and Institute of Pharmacology and Toxicology (Centro Mixto CSIC-UCM), Facultad de Medicina, Universidad Complutense, Madrid, Spain Renal ischemia/reperfusion injury (IRI) is an important cause of acute renal failure. Cellular and molecular responses of the kidney to IRI are complex and not fully understood. 1 integrins localize to the basal surface of tubular epithelium interacting with extracellular matrix components of the basal membrane, including collagen IV. Whether preservation of tubular epithelium integrity could be a therapeutic approach for IRI was assessed. The effects of HUTS-21 mAb administration, which recognizes an activation-dependent epitope of 1 integrins, in a rat model of IRI were investigated. Preischemic HUTS-21 administration resulted in the preservation of renal functional and histopathologic parameters. Analyses of activated 1 integrins expression and focal adhesion kinase phosphorylation suggest that its deactivation after IRI was prevented by HUTS-21 treatment. Moreover, HUTS-21 impaired the inflammatory response in vivo, as indicated by inhibition of proin- flammatory mediators and the absence of infiltrating cells. -

Microtubule Plus-End Tracking Proteins in Neuronal Development
Cell. Mol. Life Sci. (2016) 73:2053–2077 DOI 10.1007/s00018-016-2168-3 Cellular and Molecular Life Sciences REVIEW Microtubule plus-end tracking proteins in neuronal development 1 1 1 Dieudonne´e van de Willige • Casper C. Hoogenraad • Anna Akhmanova Received: 13 December 2015 / Revised: 4 February 2016 / Accepted: 22 February 2016 / Published online: 11 March 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Regulation of the microtubule cytoskeleton is AIS Axon initial segment of pivotal importance for neuronal development and BDNF Brain-derived neurotrophic factor function. One such regulatory mechanism centers on CAMSAP Calmodulin-regulated spectrin-associated microtubule plus-end tracking proteins (?TIPs): struc- protein turally and functionally diverse regulatory factors, which CAP-Gly Cytoskeletal-associated protein glycine-rich can form complex macromolecular assemblies at the CEP Centrosomal protein growing microtubule plus-ends. ?TIPs modulate important CFEOM1 Congenital fibrosis of the extraocular muscles properties of microtubules including their dynamics and type 1 their ability to control cell polarity, membrane transport CH Calponin homology and signaling. Several neurodevelopmental and neurode- CLASP Cytoplasmic linker protein-associated protein generative diseases are associated with mutations in ?TIPs CLIP Cytoplasmic linker protein or with misregulation of these proteins. In this review, we DRG Dorsal root ganglia focus on the role and regulation of ?TIPs in neuronal EB -

Photography and the Optical Unconscious Photography and The
PHOTOGRAPHY AND THE OPTICAL UNCONSCIOUS PHOTOGRAPHY AND THE OPTICAL UNCONSCIOUS SHAWN MICHELLE SMITH AND SHARON SLIWINSKI, EDITORS Duke University Press | Durham and London | 2017 © 2017 Duke University Press All rights reserved Printed in the United States of America on acid- free paper ♾ Cover design by Matthew Tauch; interior design by Mindy Basinger Hill Typeset in Arno Pro by Tseng Information Systems, Inc. Library of Congress Cataloging-i n-P ublication Data Names: Smith, Shawn Michelle, [date] editor. | Sliwinski, Sharon, [date] editor. Title: Photography and the optical unconscious / Shawn Michelle Smith and Sharon Sliwinski, editors. Description: Durham : Duke University Press, 2017. | Includes bibliographical references and index. Identifiers: lccn 2016048393 (print) lccn 2016050623 (ebook) isbn 9780822363811 (hardcover : alk. paper) isbn 9780822369011 (pbk. : alk. paper) isbn 9780822372998 (ebook) Subjects: lcsh: Photography—Psychological aspects. | Psychoanalysis and art. | Photography, Artistic. | Art, Modern—21st century. Classification: lcc tr183.p484 2017 (print) | lcc tr183 (ebook) | ddc 770.1—dc23 lc record available at https://lccn.loc.gov/2016048393 Cover art: Zoe Leonard, 100 North Nevill Street, 2013. Installation view, detail. Chinati Foundation, Marfa, Texas. Photo © Fredrik Nilsen, courtesy of the artist, Galerie Gisela Capitain, Cologne and Hauser & Wirth, New York. Duke University Press gratefully acknowledges the School of the Art Institute of Chicago, which provided funds toward the publication of this book. -

Transgenic Cyclooxygenase-2 Overexpression Sensitizes Mouse Skin for Carcinogenesis
Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis Karin Mu¨ ller-Decker*†, Gitta Neufang*, Irina Berger‡, Melanie Neumann*, Friedrich Marks*, and Gerhard Fu¨ rstenberger* *Research Program Tumor Cell Regulation, Deutsches Krebsforschungszentrum, and ‡Department of Pathology, Ruprecht-Karls-University, 69120 Heidelberg, Germany Edited by Philip Needleman, Pharmacia Corporation, St. Louis, MO, and approved July 29, 2002 (received for review May 30, 2002) Genetic and pharmacological evidence suggests that overexpres- there is a causal relationship between COX-2 overexpression sion of cyclooxygenase-2 (COX-2) is critical for epithelial carcino- and tumor development. Recently, we have shown that the genesis and provides a major target for cancer chemoprevention keratin 5 (K5) promoter-driven overexpression of COX-2 in by nonsteroidal antiinflammatory drugs. Transgenic mouse lines basal cells of interfollicular epidermis and the pilosebaceous unit with keratin 5 promoter-driven COX-2 overexpression in basal led to a preneoplastic skin phenotype in 4 of 4 high-expression epidermal cells exhibit a preneoplastic skin phenotype. As shown mouse lines (15). here, this phenotype depends on the level of COX-2 expression and To delineate COX-2 functions for carcinogenesis, we have COX-2-mediated prostaglandin accumulation. The transgenics did used the initiation–promotion model (2) for the induction of skin not develop skin tumors spontaneously but did so after a single tumors in wild-type (wt) NMRI mice and COX-2 transgenic application of an initiating dose of the carcinogen 7,12-dimethyl- mouse lines. This multistage model allows the analysis of the benz[a]anthracene (DMBA). Long-term treatment with the tumor carcinogenic process in terms of distinct stages, i.e., initiation by promoter phorbol 12-myristate 13-acetate, as required for tumor- application of a subcarcinogenic dose of a carcinogen such as igenesis in wild-type mice, was not necessary for transgenics. -

Keratins and Plakin Family Cytolinker Proteins Control the Length Of
RESEARCH ARTICLE Keratins and plakin family cytolinker proteins control the length of epithelial microridge protrusions Yasuko Inaba*, Vasudha Chauhan, Aaron Paul van Loon, Lamia Saiyara Choudhury, Alvaro Sagasti* Molecular, Cell and Developmental Biology Department and Molecular Biology Institute, University of California, Los Angeles, Los Angeles, United States Abstract Actin filaments and microtubules create diverse cellular protrusions, but intermediate filaments, the strongest and most stable cytoskeletal elements, are not known to directly participate in the formation of protrusions. Here we show that keratin intermediate filaments directly regulate the morphogenesis of microridges, elongated protrusions arranged in elaborate maze-like patterns on the surface of mucosal epithelial cells. We found that microridges on zebrafish skin cells contained both actin and keratin filaments. Keratin filaments stabilized microridges, and overexpressing keratins lengthened them. Envoplakin and periplakin, plakin family cytolinkers that bind F-actin and keratins, localized to microridges, and were required for their morphogenesis. Strikingly, plakin protein levels directly dictate microridge length. An actin-binding domain of periplakin was required to initiate microridge morphogenesis, whereas periplakin-keratin binding was required to elongate microridges. These findings separate microridge morphogenesis into distinct steps, expand our understanding of intermediate filament functions, and identify microridges as protrusions that integrate actin and intermediate filaments. *For correspondence: [email protected] (YI); Introduction [email protected] (AS) Cytoskeletal filaments are scaffolds for membrane protrusions that create a vast diversity of cell shapes. The three major classes of cytoskeletal elements—microtubules, actin filaments, and inter- Competing interests: The mediate filaments (IFs)—each have distinct mechanical and biochemical properties and associate authors declare that no with different regulatory proteins, suiting them to different functions. -

Hemidesmosomes and Focal Adhesions Treadmill As Separate but Linked Entities During Keratinocyte Migration Anne Pora1, Sungjun Yoon1, Reinhard Windoffer1 and Rudolf E
ORIGINAL ARTICLE See related commentary on pg 1854 Hemidesmosomes and Focal Adhesions Treadmill as Separate but Linked Entities during Keratinocyte Migration Anne Pora1, Sungjun Yoon1, Reinhard Windoffer1 and Rudolf E. Leube1 Hemidesmosomes anchor the epidermal keratin filament cytoskeleton to the extracellular matrix. They are crucial for the mechanical integrity of skin. Their role in keratinocyte migration, however, remains unclear. Examining migrating primary human keratinocytes, we find that hemidesmosomes cluster as ordered arrays consisting of multiple chevrons that are flanked by actin-associated focal adhesions. These hemidesmosomal arrays with intercalated focal adhesions extend from the cell rear to the cell front. New hemidesmosomal chevrons form subsequent to focal adhesion assembly at the cell’s leading front, whereas chevrons and associated focal adhesions disassemble at the cell rear in reverse order. The bulk of the hemidesmosome-focal adhesion composite, however, remains attached to the substratum during cell translocation. Similar hemidesmosome-focal adhesion patterns emerge on X-shaped fibronectin-coated micropatterns, during cell spreading and in leader cells during collective cell migration. We further find that hemidesmosomes and focal adhesions affect each other’s distribution. We propose that both junctions are separate but linked entities, which treadmill coordinately to support efficient directed cell migration and cooperate to coordinate the dy- namic interplay between the keratin and actin cytoskeleton. Journal of Investigative Dermatology (2019) 139, 1876e1888; doi:10.1016/j.jid.2019.03.1139 INTRODUCTION are characterized by HD-specific a6/b4 integrin hetero- Epithelial cell migration is a key process in physiological dimers, which are connected through plakins to keratins situations, such as tissue morphogenesis, and in pathological (Nahidiazar et al., 2015). -

Molecular Pathways Associated with the Nutritional Programming of Plant-Based Diet Acceptance in Rainbow Trout Following an Early Feeding Exposure Mukundh N
Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, Aurélie Le Cam, Françoise Médale, Sadasivam J. Kaushik, Inge Geurden To cite this version: Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, et al.. Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure. BMC Genomics, BioMed Central, 2016, 17 (1), pp.1-20. 10.1186/s12864-016-2804-1. hal-01346912 HAL Id: hal-01346912 https://hal.archives-ouvertes.fr/hal-01346912 Submitted on 19 Jul 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Balasubramanian et al. BMC Genomics (2016) 17:449 DOI 10.1186/s12864-016-2804-1 RESEARCH ARTICLE Open Access Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian1, Stephane Panserat1, Mathilde Dupont-Nivet2, Edwige Quillet2, Jerome Montfort3, Aurelie Le Cam3, Francoise Medale1, Sadasivam J. Kaushik1 and Inge Geurden1* Abstract Background: The achievement of sustainable feeding practices in aquaculture by reducing the reliance on wild-captured fish, via replacement of fish-based feed with plant-based feed, is impeded by the poor growth response seen in fish fed high levels of plant ingredients. -
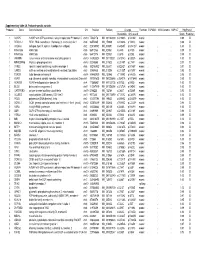
Supplementary Table 2A. Proband-Specific Variants Proband
Supplementary table 2A. Proband-specific variants Proband Gene Gene full name Chr Position RefSeqChange Function ESP6500 1000 Genome SNP137 PolyPhen2 Nucleotide Amino acid Score Prediction 1 AGAP5 ArfGAP with GTPase domain, ankyrin repeat and PH domain 5 chr10 75435178 NM_001144000 c.G1240A p.V414M exonic . 0.99 D 1 REXO1L1 REX1, RNA exonuclease 1 homolog (S. cerevisiae)-like 1 chr8 86574465 NM_172239 c.A1262G p.Y421C exonic . 0.98 D 1 COL4A3 collagen, type IV, alpha 3 (Goodpasture antigen) chr2 228169782 NM_000091 c.G4235T p.G1412V exonic . 1.00 D 1 KIAA1586 KIAA1586 chr6 56912133 NM_020931 c.C44G p.A15G exonic . 0.99 D 1 KIAA1586 KIAA1586 chr6 56912176 NM_020931 c.C87G p.D29E exonic . 0.99 D 1 JAKMIP3 Janus kinase and microtubule interacting protein 3 chr10 133955524 NM_001105521 c.G1574C p.G525A exonic . 1.00 D 1 MPHOSPH8 M-phase phosphoprotein 8 chr13 20240685 NM_017520 c.C2140T p.L714F exonic . 0.97 D 1 SYNE1 spectrin repeat containing, nuclear envelope 1 chr6 152783902 NM_033071 c.A2242T p.N748Y exonic . 0.97 D 1 CAND2 cullin-associated and neddylation-dissociated 2 (putative) chr3 12858835 NM_012298 c.C2125T p.H709Y exonic . 0.95 D 1 TDRD9 tudor domain containing 9 chr14 104460909 NM_153046 c.T1289G p.V430G exonic . 0.96 D 2 ASPM asp (abnormal spindle) homolog, microcephaly associated (Drosophila)chr1 197091625 NM_001206846 c.G3491A p.R1164H exonic . 0.99 D 2 ADAM29 ADAM metallopeptidase domain 29 chr4 175896951 NM_001130705 c.A275G p.D92G exonic . 1.00 D 2 BCO2 beta-carotene oxygenase 2 chr11 112087019 NM_001256398 c.G1373A p.G458E exonic . -

'Montalcino, a Zebrafish Model for Variegate Porphyria'
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2008 montalcino, A zebrafish model for variegate porphyria Dooley, Kimberly A ; Fraenkel, Paula G ; Langer, Nathaniel B ; Schmid, Bettina ; Davidson, Alan J ; Weber, Gerhard ; Chiang, Ken ; Foott, Helen ; Dwyer, Caitlin ; Wingert, Rebecca A ; Zhou, Yi ; Paw, Barry H ; Zon, Leonard I Abstract: OBJECTIVE Inherited or acquired mutations in the heme biosynthetic pathway leads to a debilitating class of diseases collectively known as porphyrias, with symptoms that can include anemia, cutaneous photosensitivity, and neurovisceral dysfunction. In a genetic screen for hematopoietic mutants, we isolated a zebrafish mutant, montalcino (mno), which displays hypochromic anemia and porphyria. The objective of this study was to identify the defective gene and characterize the phenotype of the zebrafish mutant. MATERIALS AND METHODS Genetic linkage analysis was utilized to identify the region harboring the mno mutation. Candidate gene analysis together with reverse transcriptase polymerase chain reaction was utilized to identify the genetic mutation, which was confirmed via allele- specific oligo hybridizations. Whole mount in situ hybridizations and o-dianisidine staining were usedto characterize the phenotype of the mno mutant. mRNA and morpholino microinjections were performed to phenocopy and/or rescue the mutant phenotype. RESULTS Homozygous mno mutant embryos have a defect in the protoporphyrinogen oxidase (ppox) gene, which encodes the enzyme that catalyzes the oxidation of protoporphyrinogen. Homozygous mutant embryos are deficient in hemoglobin, and by 36 hours post-fertilization are visibly anemic and porphyric. The hypochromic anemia of mno embryos was partially rescued by human ppox, providing evidence for the conservation of function between human and zebrafish ppox. -

Structural and Biochemical Changes Underlying a Keratoderma-Like Phenotype in Mice Lacking Suprabasal AP1 Transcription Factor Function
Citation: Cell Death and Disease (2015) 6, e1647; doi:10.1038/cddis.2015.21 OPEN & 2015 Macmillan Publishers Limited All rights reserved 2041-4889/15 www.nature.com/cddis Structural and biochemical changes underlying a keratoderma-like phenotype in mice lacking suprabasal AP1 transcription factor function EA Rorke*,1, G Adhikary2, CA Young2, RH Rice3, PM Elias4, D Crumrine4, J Meyer4, M Blumenberg5 and RL Eckert2,6,7,8 Epidermal keratinocyte differentiation on the body surface is a carefully choreographed process that leads to assembly of a barrier that is essential for life. Perturbation of keratinocyte differentiation leads to disease. Activator protein 1 (AP1) transcription factors are key controllers of this process. We have shown that inhibiting AP1 transcription factor activity in the suprabasal murine epidermis, by expression of dominant-negative c-jun (TAM67), produces a phenotype type that resembles human keratoderma. However, little is understood regarding the structural and molecular changes that drive this phenotype. In the present study we show that TAM67-positive epidermis displays altered cornified envelope, filaggrin-type keratohyalin granule, keratin filament, desmosome formation and lamellar body secretion leading to reduced barrier integrity. To understand the molecular changes underlying this process, we performed proteomic and RNA array analysis. Proteomic study of the corneocyte cross-linked proteome reveals a reduction in incorporation of cutaneous keratins, filaggrin, filaggrin2, late cornified envelope precursor proteins, hair keratins and hair keratin-associated proteins. This is coupled with increased incorporation of desmosome linker, small proline-rich, S100, transglutaminase and inflammation-associated proteins. Incorporation of most cutaneous keratins (Krt1, Krt5 and Krt10) is reduced, but incorporation of hyperproliferation-associated epidermal keratins (Krt6a, Krt6b and Krt16) is increased.