The Expression Profile and Prognostic Significance of Eukaryotic Translation Elongation Factors in Different Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

The Role of Myc-Induced Protein Synthesis in Cancer
Published OnlineFirst November 24, 2009; DOI: 10.1158/0008-5472.CAN-09-1970 Review The Role of Myc-Induced Protein Synthesis in Cancer Davide Ruggero School of Medicine and Department of Urology, Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California Abstract tions and processing by directly controlling the expression of ribo- Deregulation in different steps of translational control is an nucleases, rRNA-modifying enzymes, and nucleolar proteins NPM emerging mechanism for cancer formation. One example of involved in ribosome biogenesis such as nucleophosmin ( ), Nop52, Nop56 DKC1 an oncogene with a direct role in control of translation is , and (Table 1;ref. 16). Furthermore, Myc in- UBF the Myc transcription factor. Myc directly increases protein duces Upstream Binding Factor ( ) expression, which is an es- synthesis rates by controlling the expression of multiple com- sential transcription factor for RNA Pol I-mediated transcription ponents of the protein synthetic machinery, including ribo- (21). It has also been recently shown that a fraction of the Myc somal proteins and initiation factors of translation, Pol III protein is localized in the nucleolus and directly regulates rRNA rDNA and rDNA. However, the contribution of Myc-dependent in- synthesis by binding to E-box elements located in the pro- – creases in protein synthesis toward the multistep process moter (17 19). In addition, it can activate Pol I transcription by rDNA leading to cancer has remained unknown. Recent evidence binding and recruiting to the promoter SL1, which is essen- strongly suggests that Myc oncogenic signaling may monopo- tial for the assembly of the RNA Pol I pre-initiation complex (18). -

Supplementary Information
SUPPLEMENTARY INFORMATION Myeloperoxidase-derived 2-chlorohexadecanal is generated in mouse heart during endotoxemia and induces modification of distinct cardiomyocyte protein subsets in vitro Jürgen Prasch, Eva Bernhart, Helga Reicher, Manfred Kollroser, Gerald N. Rechberger, Chintan N. Koyani, Christopher Trummer, Lavinia Rech, Peter P. Rainer, Astrid Hammer, Ernst Malle, Wolfgang Sattler Table S1: Biological process gene ontology (GO) enrichment analysis. #term ID term description observed background false discovery matching proteins in network (labels) gene count gene count rate GO:0006457 protein folding 10 153 5.21e-09 Cct3,Cct5,Cct8,Fkbp4,Hsp90aa1,Hsp a1l,Hspb1,Pdia3,Pdia6,Tcp1 GO:0007339 binding of sperm to 6 36 4.02e-07 Aldoa,Cct3,Cct5,Cct8,Hspa1l,Tcp1 zona pellucida GO:0061077 chaperone-mediated 6 60 2.67e-06 Cct3,Cct5,Cct8,Fkbp4,Hspb1,Tcp1 protein folding GO:0017144 drug metabolic process 11 494 4.06e-06 Aldh2,Aldoa,Eno1,Gapdh,Hsp90aa1,I dh3a,Ldha,Ndufs2,Pgam1,Phgdh,Uq crc1 GO:2000573 positive regulation of 6 69 4.16e-06 Cct3,Cct5,Cct8,Ddx39b,Hsp90aa1,Tc DNA biosynthetic p1 process GO:0009987 cellular process 47 12459 4.22e-06 Alad,Alb,Aldh2,Aldoa,Cct3,Cct5,Cct8, Dctn2,Ddx39,Ddx39b,Des,Eef1g,Eef 2,Eif3f,Eif4a2,Eno1,Fdps,Fkbp4,Gap dh,Hnrnpl,Hsp90aa1,Hspa1l,Hspb1,I dh3a,Ldha,Lmna,Lyz1,Ndufs2,Pcna, Pdia3,Pdia6,Pgam1,Phgdh,Prph,Psm d13,Rpsa,Ruvbl2,Tcp1,Tuba3b,Tubal 3,Tubb3,Tubb6,Uap1l1,Uqcrc1,Uqcrc 2,Vim,Ywhab GO:1904851 positive regulation of 4 10 4.22e-06 Cct3,Cct5,Cct8,Tcp1 establishment of protein localization to telomere GO:0046031 -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Disease-Related Cellular Protein Networks Differentially Affected
www.nature.com/scientificreports OPEN Disease‑related cellular protein networks diferentially afected under diferent EGFR mutations in lung adenocarcinoma Toshihide Nishimura1,8*, Haruhiko Nakamura1,2,8, Ayako Yachie3,8, Takeshi Hase3,8, Kiyonaga Fujii1,8, Hirotaka Koizumi4, Saeko Naruki4, Masayuki Takagi4, Yukiko Matsuoka3, Naoki Furuya5, Harubumi Kato6,7 & Hisashi Saji2 It is unclear how epidermal growth factor receptor EGFR major driver mutations (L858R or Ex19del) afect downstream molecular networks and pathways. This study aimed to provide information on the infuences of these mutations. The study assessed 36 protein expression profles of lung adenocarcinoma (Ex19del, nine; L858R, nine; no Ex19del/L858R, 18). Weighted gene co-expression network analysis together with analysis of variance-based screening identifed 13 co-expressed modules and their eigen proteins. Pathway enrichment analysis for the Ex19del mutation demonstrated involvement of SUMOylation, epithelial and mesenchymal transition, ERK/mitogen- activated protein kinase signalling via phosphorylation and Hippo signalling. Additionally, analysis for the L858R mutation identifed various pathways related to cancer cell survival and death. With regard to the Ex19del mutation, ROCK, RPS6KA1, ARF1, IL2RA and several ErbB pathways were upregulated, whereas AURK and GSKIP were downregulated. With regard to the L858R mutation, RB1, TSC22D3 and DOCK1 were downregulated, whereas various networks, including VEGFA, were moderately upregulated. In all mutation types, CD80/CD86 (B7), MHC, CIITA and IFGN were activated, whereas CD37 and SAFB were inhibited. Costimulatory immune-checkpoint pathways by B7/CD28 were mainly activated, whereas those by PD-1/PD-L1 were inhibited. Our fndings may help identify potential therapeutic targets and develop therapeutic strategies to improve patient outcomes. -
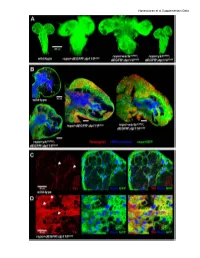
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Review EEF1G (Eukaryotic translation elongation factor 1 gamma) Luigi Cristiano Aesthetic and medical biotechnologies research unit, Prestige, Terranuova Bracciolini, Italy; [email protected] Published in Atlas Database: March 2019 Online updated version : http://AtlasGeneticsOncology.org/Genes/EEF1GID54272ch11q12.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/70656/03-2019-EEF1GID54272ch11q12.pdf DOI: 10.4267/2042/70656 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2020 Atlas of Genetics and Cytogenetics in Oncology and Haematology Abstract Keywords EEF1G; Eukaryotic translation elongation factor 1 Eukaryotic translation elongation factor 1 gamma, gamma; Translation; Translation elongation factor; alias eEF1G, is a protein that plays a main function protein synthesis; cancer; oncogene; cancer marker in the elongation step of translation process but also covers numerous moonlighting roles. Considering its Identity importance in the cell it is found frequently Other names: EF1G, GIG35, PRO1608, EEF1γ, overexpressed in human cancer cells and thus this EEF1Bγ review wants to collect the state of the art about EEF1G, with insights on DNA, RNA, protein HGNC (Hugo): EEF1G encoded and the diseases where it is implicated. Location: 11q12.3 Figure. 1. Splice variants of EEF1G. The figure shows the locus on chromosome 11 of the EEF1G gene and its splicing variants (grey/blue box). The primary transcript is EEF1G-001 mRNA (green/red box), but also EEF1G-201 variant is able to codify for a protein (reworked from https://www.ncbi.nlm.nih.gov/gene/1937; http://grch37.ensembl.org; www.genecards.org) Atlas Genet Cytogenet Oncol Haematol. -

Global Analysis of LARP1 Translation Targets Reveals Tunable and Dynamic Features of 5′ TOP Motifs
Global analysis of LARP1 translation targets reveals tunable and dynamic features of 5′ TOP motifs Lucas Philippea,1, Antonia M. G. van den Elzena,1, Maegan J. Watsona, and Carson C. Thoreena,2 aDepartment of Cellular and Molecular Physiology, Yale School of Medicine, New Haven, CT 06510 Edited by Alan G. Hinnebusch, National Institutes of Health, Bethesda, MD, and approved January 29, 2020 (received for review July 25, 2019) Terminal oligopyrimidine (TOP) motifs are sequences at the 5′ ends recent findings have hinted that the RNA-binding protein La- of mRNAs that link their translation to the mTOR Complex 1 related protein 1 (LARP1) may have a central role (8–10). (mTORC1) nutrient-sensing signaling pathway. They are com- LARP1 is a large protein (150 kDa) with several RNA-binding monly regarded as discrete elements that reside on ∼100 mRNAs domains. Its central region contains a La motif (LaM) domain that mostly encode translation factors. However, the full spectrum that defines the La-related protein (LARP) superfamily, along of TOP sequences and their prevalence throughout the transcrip- with an adjacent RNA recognition motif-like (RRM-L) domain. tome remain unclear, primarily because of uncertainty over the Its C terminus encodes a domain known as the DM15 region. This mechanism that detects them. Here, we globally analyzed trans- domain is unique to LARP1 and its closely related homolog lation targets of La-related protein 1 (LARP1), an RNA-binding pro- LARP1B, and is therefore also known as the LARP1 domain (11). tein and mTORC1 effector that has been shown to repress TOP Several observations suggest that LARP1 directly represses TOP mRNA translation in a few specific cases. -

Comparative Transcriptome Analysis Reveals Different Host Cell Responses to Acute and Persistent Foot-And-Mouth Disease Virus Infection
Virologica Sinica www.virosin.org https://doi.org/10.1007/s12250-019-00155-8 www.springer.com/12250 (0123456789().,-volV)(0123456789().,-volV) RESEARCH ARTICLE Comparative Transcriptome Analysis Reveals Different Host Cell Responses to Acute and Persistent Foot-and-Mouth Disease Virus Infection 1 1 1 1 1 1 1 Jiadai Li • Lingling Han • Yao Hao • Yuncong Yuan • Mingzhen Wang • Xiu Xin • Hailong Wang • 2 1,3 1,3 Fang Yu • Congyi Zheng • Chao Shen Received: 15 March 2019 / Accepted: 3 July 2019 Ó Wuhan Institute of Virology, CAS 2019 Abstract Foot-and-mouth disease virus (FMDV) rapidly causes cytopathic effects in susceptible cells. Incomplete viral clearance during the acute infection leads to persistent infection. The relationship between host gene expression and the persistent infection remains unclear. In this study, we analyzed the transcriptome profiles of BHK-21 cells acutely and persistently infected with FMDV to identify differences in gene expression. GO and KEGG enrichment analyses showed that the 8,378 differentially expressed genes were significantly enriched in categories including metabolism, biosynthesis, ribosome function, and endocytosis. In persistently infected BHK-21 cells, ribosome- and translation-related genes were significantly down-regulated. There were more differentially expressed immune-related genes during persistent infection than during acute infection. Two hundred and seventy-four genes were differentially expressed in both acutely and persistently infected BHK-21 cells. Among these genes, heat shock protein family B member 1 (Hspb1) knockdown significantly inhibited FMDV replication. Our research provides a basis for further research to understand the mechanisms of persistent FMDV infection including the genes involved in FMDV replication. -

Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’S Disease Lavida R.K
bioRxiv preprint doi: https://doi.org/10.1101/498527; this version posted December 17, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’s Disease Lavida R.K. Brooks 1, George I. Mias,2;∗ 1 Institute for Quantitative Health Science and Engineering, Michigan State University, Microbiology and Molecular Genetics, East Lansing, 48824, USA 2 Institute for Quantitative Health Science and Engineering, Michigan State University, Biochemistry and Molecular Biology, East Lansing, 48824, USA Correspondence*: George I. Mias [email protected] 2 ABSTRACT 3 Alzheimer’s disease (AD) has been categorized by the Centers for Disease Control and 4 Prevention (CDC) as the 6th leading cause of death in the United States. AD is a significant 5 health-care burden because of its increased occurrence (specifically in the elderly population) 6 and the lack of effective treatments and preventive methods. With an increase in life expectancy, 7 the CDC expects AD cases to rise to 15 million by 2060. Aging has been previously associated 8 with susceptibility to AD, and there are ongoing efforts to effectively differentiate between normal 9 and AD age-related brain degeneration and memory loss. AD targets neuronal function and can 10 cause neuronal loss due to the buildup of amyloid-beta plaques and intracellular neurofibrillary 11 tangles. 12 Our study aims to identify temporal changes within gene expression profiles of healthy controls 13 and AD subjects. -

Global Analysis of Trna and Translation Factor Expression Reveals a Dynamic Landscape of Translational Regulation in Human Cancers
ARTICLE https://doi.org/10.1038/s42003-018-0239-8 OPEN Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers Zhao Zhang1, Youqiong Ye1, Jing Gong1, Hang Ruan1, Chun-Jie Liu2, Yu Xiang1, Chunyan Cai3, An-Yuan Guo2, 1234567890():,; Jiqiang Ling4, Lixia Diao5, John N. Weinstein5 & Leng Han 1,6 The protein translational system, including transfer RNAs (tRNAs) and several categories of enzymes, plays a key role in regulating cell proliferation. Translation dysregulation also contributes to cancer development, though relatively little is known about the changes that occur to the translational system in cancer. Here, we present global analyses of tRNAs and three categories of enzymes involved in translational regulation in ~10,000 cancer patients across 31 cancer types from The Cancer Genome Atlas. By analyzing the expression levels of tRNAs at the gene, codon, and amino acid levels, we identified unequal alterations in tRNA expression, likely due to the uneven distribution of tRNAs decoding different codons. We find that overexpression of tRNAs recognizing codons with a low observed-over-expected ratio may overcome the translational bottleneck in tumorigenesis. We further observed overall overexpression and amplification of tRNA modification enzymes, aminoacyl-tRNA synthe- tases, and translation factors, which may play synergistic roles with overexpression of tRNAs to activate the translational systems across multiple cancer types. 1 Department of Biochemistry and Molecular Biology, McGovern Medical School at The University of Texas Health Science Center at Houston, Houston, TX 77030, USA. 2 Department of Bioinformatics and Systems Biology, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology Wuhan, 430074 Hubei, People’s Republic of China. -

And/Or HCV-Associated Hepatocellular Carcinoma Cells Using Expressed Sequence Tags
315-327 28/6/06 12:53 Page 315 INTERNATIONAL JOURNAL OF ONCOLOGY 29: 315-327, 2006 315 Gene expression profiling of human HBV- and/or HCV-associated hepatocellular carcinoma cells using expressed sequence tags SUN YOUNG YOON1, JEONG-MIN KIM1, JUNG-HWA OH1, YEO-JIN JEON1, DONG-SEOK LEE1, JOO HEON KIM3, JONG YOUNG CHOI4, BYUNG MIN AHN5, SANGSOO KIM2, HYANG-SOOK YOO1, YONG SUNG KIM1 and NAM-SOON KIM1 1Laboratory of Human Genomics, Genome Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon 305-333; 2Department of Bioinformatics and Life Science, Soongsil University, Seoul; 3Department of Pathology, Eulji University School of Medicine, Daejeon 301-832; 4Department of Internal Medicine, Catholic University of Medicine, 137-701 Seoul; 5Department of Internal Medicine, Catholic University of Medicine, Daejeon 301-723, Korea Received November 14, 2005; Accepted January 19, 2006 Abstract. Liver cancer is one of the leading causes of cancer expressed at high levels in HCC. Using an analysis of EST death worldwide. To identify novel target genes that are frequency, the newly identified genes, especially ANXA2, related to liver carcinogenesis, we examined new genes represent potential biomarkers for HCC and useful targets for that are differentially expressed in human hepatocellular elucidating the molecular mechanisms associated with HCC carcinoma (HCC) cell lines and tissues based on the expressed involving virological etiology. sequence tag (EST) frequency. Eleven libraries were constructed from seven HCC cell lines and three normal liver Introduction tissue samples obtained from Korean patients. An analysis of gene expression profiles for HCC was performed using the Liver cancer is one of the leading causes of cancer death frequency of ESTs obtained from these cDNA libraries.