The Revised Ghent Nosology; Reclassifying Isolated Ectopia Lentis A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ocular Surface Changes Associated with Ophthalmic Surgery
Journal of Clinical Medicine Review Ocular Surface Changes Associated with Ophthalmic Surgery Lina Mikalauskiene 1, Andrzej Grzybowski 2,3 and Reda Zemaitiene 1,* 1 Department of Ophthalmology, Medical Academy, Lithuanian University of Health Sciences, 44037 Kaunas, Lithuania; [email protected] 2 Department of Ophthalmology, University of Warmia and Mazury, 10719 Olsztyn, Poland; [email protected] 3 Institute for Research in Ophthalmology, Foundation for Ophthalmology Development, 61553 Poznan, Poland * Correspondence: [email protected] Abstract: Dry eye disease causes ocular discomfort and visual disturbances. Older adults are at a higher risk of developing dry eye disease as well as needing for ophthalmic surgery. Anterior segment surgery may induce or worsen existing dry eye symptoms usually for a short-term period. Despite good visual outcomes, ocular surface dysfunction can significantly affect quality of life and, therefore, lower a patient’s satisfaction with ophthalmic surgery. Preoperative dry eye disease, factors during surgery and postoperative treatment may all contribute to ocular surface dysfunction and its severity. We reviewed relevant articles from 2010 through to 2021 using keywords “cataract surgery”, ”phacoemulsification”, ”refractive surgery”, ”trabeculectomy”, ”vitrectomy” in combina- tion with ”ocular surface dysfunction”, “dry eye disease”, and analyzed studies on dry eye disease pathophysiology and the impact of anterior segment surgery on the ocular surface. Keywords: dry eye disease; ocular surface dysfunction; cataract surgery; phacoemulsification; refractive surgery; trabeculectomy; vitrectomy Citation: Mikalauskiene, L.; Grzybowski, A.; Zemaitiene, R. Ocular Surface Changes Associated with Ophthalmic Surgery. J. Clin. 1. Introduction Med. 2021, 10, 1642. https://doi.org/ 10.3390/jcm10081642 Dry eye disease (DED) is a common condition, which usually causes discomfort, but it can also be an origin of ocular pain and visual disturbances. -

BOSTON TERRIER EYE DISEASE Corneal Ulcers and Prevention
BOSTON TERRIER EYE DISEASE Corneal Ulcers and Prevention Corneal Ulcers are the single largest eye problem in Boston Terriers. Perhaps 1 dog in 10 will experience a corneal ulcer sometime during its life based on the l903 dogs surveyed in the 2000 Boston Terrier Health Survey. The Boston Terrier Standard for the Breed calls for eyes to be “wide apart, large and round and dark in color. The eyes are set square in the skull and the outside corners are on a line with the cheeks as viewed from the front". The ideal Boston Terrier eye does not protrude but is "set square in the skull". Unfortunately the Boston eye is fairly prone to eye injury because of its large size and prominence. Corneal ulcers are caused initially by injury to the eyes. The common practice of removing Boston Terrier whiskers may be a reason that eyes become injured due to lack of sensory feelers. Some breeders do not trim whiskers once a dog's show career is finished because they know that whiskers can prevent injury to the eye. There are a number of external reasons why an injured eye doesn't heal. These may include irritation from eyelashes or from facial hairs, infection, and lack of moisture in the eye. Some of these reasons are hereditary. Internal reasons for an eye not healing include glaucoma and infection. Corneal ulcers can be difficult and expensive to treat and often result in the loss of the eye. This is a case where an "ounce of prevention is worth a pound of cure". -

Causes of Heterochromia Iridis with Special Reference to Paralysis Of
CAUSES OF HETEROCHROMIA IRIDIS WITH SPECIAL REFER- ENCE TO PARALYSIS OF THE CERVICAL SYMPATHETIC. F. PHINIZY CALHOUN, M. D. ATLANTA, GA. This abstract of a candidate's thesis presented for membership in the American Ophthal- mological Society, includes the reports of cases, a general review of the literature of the sub- ject, the results of experiments, and histologic observations on the effect of extirpation of the cervical sympathetic in the rab'bit, the conclusions reached from the investigation, and a bib- liography. That curious condition which con- thinks that the word hetcrochromia sists in a difference in the pigmentation should apply to those cases in which of the two eyes, is regarded by the parts of the same iris have different casual observer as a play or caprice of colors. In those cases where a cycli- nature. This phenomenon has for cen- tis accompanies the iris decoloration, turies been noted, and was called hcte- Butler8 uses the term "heterochromic roglaucus by Aristotle1. One who cyclitis," but the "Chronic Cyclitis seriously studies the subject, is at once with Decoloration of the Iris" as de- impressed with the complexity of the scribed by Fuchs" undoubtedly gives a situation, and soon learns that nature more accurate description of the dis- plays a comparatively small part in its ease, notwithstanding its long title. causation. It is however only within The commonly accepted and most uni- a comparatively recent time that the versally used term Hetcrochromia Iri- pathologic aspect has been considered, dis exactly expresses and implies the and in this discussion I especially wish picture from its derivation (irtpoa to draw attention to that part played other, xpw/xa) color. -

Retinitis Pigmentosa Type 11 - a USD1- 2B P.A
Corporate Life-changing science Presentation February 2021 Overview PYC is an RNA therapeutics company with an initial focus on diseases of the eye § RNA therapeutics have come of age But their ongoing success is impeded by inefficient or toxic delivery inside cells § PYC’s cell-penetrating peptide (CPP) delivery platform solves this ‘delivery’ problem PYC’s competitive advantage is getting more drug safely into the target cell § PYC is applying this advantage to develop drugs for eye disease: an area of unmet need PYC’s lead program is the first disease-modifying therapy for Retinitis Pigmentosa type 11 - a USD1- 2B p.a. target market § PYC’s technology scales rapidly in the eye: same delivery tech for other RNA cargoes PYC has two other defined drug programs, each with blockbuster potential, addressing Diabetic Retinopathy and Autosomal Dominant Optic Atrophy § Building on its success in the eye, PYC is expanding the application of its technology The Company’s initial focus outside the eye is on neurodegenerative diseases 2 Corporate Snapshot (ASX: PYC) Financial Information (29 January 2021, AUD) Share Price Performance (12 months) ASX website Share price $0.14 Number of shares 3,170M Market Capitalisation $445M Cash $57M Debt Nil Enterprise Value $388M Board of Directors Alan Tribe – Chairman Sahm Nasseri– Chief Executive Officer (USA) Dr Rohan Hockings – Chief Executive Officer (Australia) Building out a US base to complement Australian discovery hub Dr Bernard Hockings – Non-Executive Director • Early discovery and candidate proof -

Brittle Cornea, Blue Sclera, and Red Hair Syndrome (The Brittle Cornea Syndrome)
Br J Ophthalmol: first published as 10.1136/bjo.64.3.175 on 1 March 1980. Downloaded from British Journal of Ophthalmology, 1980, 64, 175-177 Brittle cornea, blue sclera, and red hair syndrome (the brittle cornea syndrome) U. TICHO, M. IVRY, AND S. MERIN From the Department of Ophthalmology, Hadassah University Hospital, Jerusalem, Israel SUMVMARY A syndrome of red hair, blue sclera, and brittle cornea with recurrent spontaneous p,!fforations is presented in 2 siblings of a Tunisian Jewish family. The genetic transmission of this disorder is autosomal recessive. This is the second description of this syndrome, which should be called the 'brittle cornea syndrome'. This syndrome has so far been reported only in Tunisian Jewish families. Brittle cornea with spontaneous perforation is a visual acuity of 6/60 with the best myopic correc- rare disease. It has been described in association tion. The cornea was extremely thin with kerato- with systemic mesodermal disorders such as osteo- globus (Fig. 3). The anterior chamber was deep genesis imrperfecta,' Marfan syndrome,2 and Ehlers- and the lens was clear. No abnormalities were Danlos syndrome.3 present in the posterior segment. The intraocular This communication describes a 'brittle cornea' pressure was 16 mmHg. syndrome unrelated to systemic mesodermal disor- Examination of the left eye revealed a visual ders. The triad of symptoms includes red hair, acuity of counting fingers at 2 meters. A large brittle megalocornea, and blue sclera. The following corneal scar was present in the centre of the left 2 cases and the 4 other cases reported previously4 cornea with anterior iris adhesions (Fig. -

Targeted Deletion of Fibrillin-1 in the Mouse Eye Results in Ectopia Lentis and Other Ocular Phenotypes Associated with Marfan Syndrome Wendell Jones
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome Wendell Jones Juan Rodriguez Steven Bassnett Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs © 2019. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2019) 12, dmm037283. doi:10.1242/dmm.037283 RESEARCH ARTICLE Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome Wendell Jones1,*, Juan Rodriguez2 and Steven Bassnett1 ABSTRACT the disease nosology (the other being aortic root dilation) (Loeys Fibrillin is an evolutionarily ancient protein that lends elasticity and et al., 2010). resiliency to a variety of tissues. In humans, mutations in fibrillin-1 To investigate the roles of FBN1 mutations in the pathophysiology cause Marfan and related syndromes, conditions in which the eye is of MFS and related conditions, the Fbn1 locus has been targeted often severely affected. To gain insights into the ocular sequelae of extensively in mice. Numerous models have been generated, Marfan syndrome, we targeted Fbn1 in mouse lens or non-pigmented including hypomorphs, nulls and missense mutations (Sakai et al., ciliary epithelium (NPCE). Conditional knockout of Fbn1 in NPCE, but 2016). Collectively, these capture key features of human MFS, not lens, profoundly affected the ciliary zonule, the system of fibrillin- including kyphoscoliosis, rib overgrowth (Judge et al., 2004) and rich fibers that centers the lens in the eye. The tensile strength of the aneurysms of the ascending aorta (Pereira et al., 1997). -

Mutational Analysis of PAX6: 16 Novel Mutations Including 5 Missense Mutations with a Mild Aniridia Phenotype
European Journal of Human Genetics (1999) 7, 274–286 t © 1999 Stockton Press All rights reserved 1018–4813/99 $12.00 http://www.stockton-press.co.uk/ejhg ARTICLE Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype Karen Grønskov1, Thomas Rosenberg2, Annie Sand1 and Karen Brøndum-Nielsen1 1Department of Medical Genetics, John F Kennedy Institute, Glostrup 2National Eye Clinic for the Visually Impaired, Hellerup, Denmark Mutations in the developmental control gene PAX6 have been shown to be the genetic cause of aniridia, which is a severe panocular eye disease characterised by iris hypoplasia. The inheritance is autosomal dominant with high penetrance but variable expressivity. Here we describe a mutational analysis of 27 Danish patients using a dideoxy fingerprinting method, which identified PAX6 mutations in 18 individuals with aniridia. A thorough phenotype description was made for the 18 patients. A total of 19 mutations, of which 16 were novel, are described. Among these were five missense mutations which tended to be associated with a milder aniridia phenotype, and in fact one of them seemed to be non-penetrant. Four of the five missense mutations were located in the paired domain. We also describe a third alternative spliced PAX6 isoform in which two of the four missense mutations would be spliced out. Our observations support the concept of dosage effects of PAX6 mutations as well as presenting evidence for variable expressivity and gonadal mosaicism. Keywords: PAX6; aniridia; phenotype–genotype correlation; missense mutations; alternative splicing Introduction but variable expressivity.1 The sporadic cases can be part of the WAGR syndrome (Wilms’ tumour, aniridia, Aniridia is a severe panocular disorder, with a reported genitourinary abnormalities and mental retardation) incidence of 1 in 50 000 to 100 000. -

Upper Eyelid Ptosis Revisited Padmaja Sudhakar, MBBS, DNB (Ophthalmology) Qui Vu, BS, M3 Omofolasade Kosoko-Lasaki, MD, MSPH, MBA Millicent Palmer, MD
® AmericAn JournAl of clinicAl medicine • Summer 2009 • Volume Six, number Three 5 Upper Eyelid Ptosis Revisited Padmaja Sudhakar, MBBS, DNB (Ophthalmology) Qui Vu, BS, M3 Omofolasade Kosoko-Lasaki, MD, MSPH, MBA Millicent Palmer, MD Abstract Epidemiology of Ptosis Blepharoptosis, commonly referred to as ptosis is an abnormal Although ptosis is commonly encountered in patients of all drooping of the upper eyelid. This condition has multiple eti- ages, there are insufficient statistics regarding the prevalence ologies and is seen in all age groups. Ptosis results from a con- and incidence of ptosis in the United States and globally.2 genital or acquired weakness of the levator palpebrae superioris There is no known ethnic or sexual predilection.2 However, and the Muller’s muscle responsible for raising the eyelid, dam- there have been few isolated studies on the epidemiology of age to the nerves which control those muscles, or laxity of the ptosis. A study conducted by Baiyeroju et al, in a school and a skin of the upper eyelids. Ptosis may be found isolated, or may private clinic in Nigeria, examined 25 cases of blepharoptosis signal the presence of a more serious underlying neurological and found during a five-year period that 52% of patients were disorder. Treatment depends on the underlying etiology. This less than 16 years of age, while only 8% were over 50 years review attempts to give an overview of ptosis for the primary of age. There was a 1:1 male to female ratio in the study with healthcare provider with particular emphasis on the classifica- the majority (68%) having only one eye affected. -

A Review of Acquired Blepharoptosis: Prevalence, Diagnosis, and Current Treatment Options
Eye (2021) 35:2468–2481 https://doi.org/10.1038/s41433-021-01547-5 REVIEW ARTICLE A review of acquired blepharoptosis: prevalence, diagnosis, and current treatment options 1 2 3 4 Jason Bacharach ● Wendy W. Lee ● Andrew R. Harrison ● Thomas F. Freddo Received: 11 February 2021 / Revised: 15 March 2021 / Accepted: 7 April 2021 / Published online: 29 April 2021 © The Author(s) 2021. This article is published with open access Abstract Blepharoptosis (ptosis) is among the most common disorders of the upper eyelid encountered in both optometric and ophthalmic practice. The unilateral or bilateral drooping of the upper eyelid that characterises ptosis can affect appearance and impair visual function, both of which can negatively impact quality of life. While there are several known forms of congenital ptosis, acquired ptosis (appearing later in life, due to a variety of causes) is the predominant form of the condition. This review summarises the prevalence, causes, identification, differential diagnosis, and treatment of acquired ptosis. Particular attention is paid to the differential diagnosis of acquired ptosis and emerging treatment options, including surgical and pharmacologic approaches. 1234567890();,: 1234567890();,: Literature search notes either congenital (present at or shortly following birth) or acquired (appearing later in life). Ptosis is broadly recog- Literature cited in this review was identified via a broad nised as being among the most common disorders of the search of the PUBMED online database for English-lan- eyelid encountered -
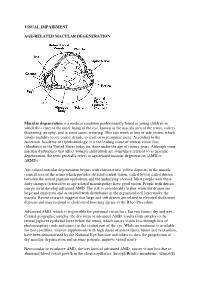
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

Clinical Signs of Brachycephalic Ocular Syndrome in 93 Dogs Joana Costa1* , Andrea Steinmetz2 and Esmeralda Delgado3
Costa et al. Irish Veterinary Journal (2021) 74:3 https://doi.org/10.1186/s13620-021-00183-5 RESEARCH Open Access Clinical signs of brachycephalic ocular syndrome in 93 dogs Joana Costa1* , Andrea Steinmetz2 and Esmeralda Delgado3 Abstract Background: Brachycephalic breeds have anatomical skull changes that are responsible for ocular clinical signs, known as the brachycephalic ocular syndrome (BOS). Their popularity has increased in recent years but the excessive pressure of selection lead to extreme conformation of skull shapes, resulting in facial alterations that can put these dogs’ vision at risk. Objectives: This study aimed to analyse the ocular disorders in a sample of 93 brachycephalic dogs to better characterize the disease complex BOS. Material and methods: Brachycephalic dogs were submitted to a complete ophthalmological examination. The studied parameters included animal’s sex, age and breed, age, ophthalmological tests performed, results of complementary exams, clinical signs, ocular disorders, treatment protocols and their outcomes. Data were organized using Microsoft Office Excel 2007® and statistical analysis was performed with IBM SPSS Statistics 20®. Results: The studied population included 93 brachycephalic dogs 45 males (48%) and 48 females (52%) from different breeds: French Bulldog (n = 38), Shih-Tzu (n = 22), Pug (n = 17), English Bulldog (n = 5), Pekingese (n = 4), Boxer (n = 4) and Boston Terrier (n = 3), aged between 0.2–16 years, median 4.65 years. The most frequent ocular abnormalities were corneal ulcers in 44%, corneal pigmentation in 36%, corneal fibrosis in 25% and entropion in 22% of the animals. There was a higher incidence of corneal pigmentary keratitis in Pugs (53%) and corneal fibrosis in Shih Tzus (36%). -

Cornea/External Disease 2017-2019
Academy MOC Essentials® Practicing Ophthalmologists Curriculum 2017–2019 Cornea/External Disease *** Cornea/External Disease 2 © AAO 2017-2019 Practicing Ophthalmologists Curriculum Disclaimer and Limitation of Liability As a service to its members and American Board of Ophthalmology (ABO) diplomates, the American Academy of Ophthalmology has developed the Practicing Ophthalmologists Curriculum (POC) as a tool for members to prepare for the Maintenance of Certification (MOC) -related examinations. The Academy provides this material for educational purposes only. The POC should not be deemed inclusive of all proper methods of care or exclusive of other methods of care reasonably directed at obtaining the best results. The physician must make the ultimate judgment about the propriety of the care of a particular patient in light of all the circumstances presented by that patient. The Academy specifically disclaims any and all liability for injury or other damages of any kind, from negligence or otherwise, for any and all claims that may arise out of the use of any information contained herein. References to certain drugs, instruments, and other products in the POC are made for illustrative purposes only and are not intended to constitute an endorsement of such. Such material may include information on applications that are not considered community standard, that reflect indications not included in approved FDA labeling, or that are approved for use only in restricted research settings. The FDA has stated that it is the responsibility of the physician to determine the FDA status of each drug or device he or she wishes to use, and to use them with appropriate patient consent in compliance with applicable law.