Practical Linux Examples
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

At—At, Batch—Execute Commands at a Later Time
at—at, batch—execute commands at a later time at [–csm] [–f script] [–qqueue] time [date] [+ increment] at –l [ job...] at –r job... batch at and batch read commands from standard input to be executed at a later time. at allows you to specify when the commands should be executed, while jobs queued with batch will execute when system load level permits. Executes commands read from stdin or a file at some later time. Unless redirected, the output is mailed to the user. Example A.1 1 at 6:30am Dec 12 < program 2 at noon tomorrow < program 3 at 1945 pm August 9 < program 4 at now + 3 hours < program 5 at 8:30am Jan 4 < program 6 at -r 83883555320.a EXPLANATION 1. At 6:30 in the morning on December 12th, start the job. 2. At noon tomorrow start the job. 3. At 7:45 in the evening on August 9th, start the job. 4. In three hours start the job. 5. At 8:30 in the morning of January 4th, start the job. 6. Removes previously scheduled job 83883555320.a. awk—pattern scanning and processing language awk [ –fprogram–file ] [ –Fc ] [ prog ] [ parameters ] [ filename...] awk scans each input filename for lines that match any of a set of patterns specified in prog. Example A.2 1 awk '{print $1, $2}' file 2 awk '/John/{print $3, $4}' file 3 awk -F: '{print $3}' /etc/passwd 4 date | awk '{print $6}' EXPLANATION 1. Prints the first two fields of file where fields are separated by whitespace. 2. Prints fields 3 and 4 if the pattern John is found. -

Application for a Certificate of Eligibility to Employ Child Performers
Division of Labor Standards Permit and Certificate Unit Harriman State Office Campus Building 12, Room 185B Albany, NY 12240 www.labor.ny.gov Application for a Certificate of Eligibility to Employ Child Performers A. Submission Instructions A Certificate of Eligibility to Employ Child Performers must be obtained prior to employing any child performer. Certificates are renew able every three (3) years. To obtain or renew a certificate: • Complete Parts B, C, D and E of this application. • Attach proof of New York State Workers’ Compensation and Disability Insurance. o If you currently have employees in New York, you must provide proof of coverage for those New York State w orkers by attaching copies of Form C-105.2 and DB-120.1, obtainable from your insurance carrier. o If you are currently exempt from this requirement, complete Form CE-200 attesting that you are not required to obtain New York State Workers’ Compensation and Disability Insurance Coverage. Information on and copies of this form are available from any district office of the Workers’ Compensation Board or from their w ebsite at w ww.wcb.ny.gov, Click on “WC/DB Exemptions,” then click on “Request for WC/DB Exemptions.” • Attach a check for the correct amount from Section D, made payable to the Commissioner of Labor. • Sign and mail this completed application and all required documents to the address listed above. If you have any questions, call (518) 457-1942, email [email protected] or visit the Department’s w ebsite at w ww.labor.ny.gov B. Type of Request (check one) New Renew al Current certificate number _______________________________________________ Are you seeking this certificate to employ child models? Yes No C. -

Program #6: Word Count
CSc 227 — Program Design and Development Spring 2014 (McCann) http://www.cs.arizona.edu/classes/cs227/spring14/ Program #6: Word Count Due Date: March 11 th, 2014, at 9:00 p.m. MST Overview: The UNIX operating system (and its variants, of which Linux is one) includes quite a few useful utility programs. One of those is wc, which is short for Word Count. The purpose of wc is to give users an easy way to determine the size of a text file in terms of the number of lines, words, and bytes it contains. (It can do a bit more, but that’s all of the functionality that we are concerned with for this assignment.) Counting lines is done by looking for “end of line” characters (\n (ASCII 10) for UNIX text files, or the pair \r\n (ASCII 13 and 10) for Windows/DOS text files). Counting words is also straight–forward: Any sequence of characters not interrupted by “whitespace” (spaces, tabs, end–of–line characters) is a word. Of course, whitespace characters are characters, and need to be counted as such. A problem with wc is that it generates a very minimal output format. Here’s an example of what wc produces on a Linux system when asked to count the content of a pair of files; we can do better! $ wc prog6a.dat prog6b.dat 2 6 38 prog6a.dat 32 321 1883 prog6b.dat 34 327 1921 total Assignment: Write a Java program (completely documented according to the class documentation guidelines, of course) that counts lines, words, and bytes (characters) of text files. -

Course Outline & Schedule
Course Outline & Schedule Call US 408-759-5074 or UK +44 20 7620 0033 Linux Advanced Shell Programming Tools Curriculum The Linux Foundation Course Code LASP Duration 3 Day Course Price $1,830 Course Description The Linux Advanced Shell Programming Tools course is designed to give delegates practical experience using a range of Linux tools to manipulate text and incorporate them into Linux shell scripts. The delegate will practise: Using the shell command line editor Backing up and restoring files Scheduling background jobs using cron and at Using regular expressions Editing text files with sed Using file comparison utilities Using the head and tail utilities Using the cut and paste utilities Using split and csplit Identifying and translating characters Sorting files Translating characters in a file Selecting text from files with the grep family of commands Creating programs with awk Course Modules Review of Shell Fundamentals (1 topic) ◾ Review of UNIX Commands Using Unix Shells (6 topics) ◾ Command line history and editing ◾ The Korn and POSIX shells Perpetual Solutions - Page 1 of 5 Course Outline & Schedule Call US 408-759-5074 or UK +44 20 7620 0033 ◾ The Bash shell ◾ Command aliasing ◾ The shell startup file ◾ Shell environment variables Redirection, Pipes and Filters (7 topics) ◾ Standard I/O and redirection ◾ Pipes ◾ Command separation ◾ Conditional execution ◾ Grouping Commands ◾ UNIX filters ◾ The tee command Backup and Restore Utilities (6 topics) ◾ Archive devices ◾ The cpio command ◾ The tar command ◾ The dd command ◾ Exercise: -

HEP Computing Part I Intro to UNIX/LINUX Adrian Bevan
HEP Computing Part I Intro to UNIX/LINUX Adrian Bevan Lectures 1,2,3 [email protected] 1 Lecture 1 • Files and directories. • Introduce a number of simple UNIX commands for manipulation of files and directories. • communicating with remote machines [email protected] 2 What is LINUX • LINUX is the operating system (OS) kernel. • Sitting on top of the LINUX OS are a lot of utilities that help you do stuff. • You get a ‘LINUX distribution’ installed on your desktop/laptop. This is a sloppy way of saying you get the OS bundled with lots of useful utilities/applications. • Use LINUX to mean anything from the OS to the distribution we are using. • UNIX is an operating system that is very similar to LINUX (same command names, sometimes slightly different functionalities of commands etc). – There are usually enough subtle differences between LINUX and UNIX versions to keep you on your toes (e.g. Solaris and LINUX) when running applications on multiple platforms …be mindful of this if you use other UNIX flavours. – Mac OS X is based on a UNIX distribution. [email protected] 3 Accessing a machine • You need a user account you should all have one by now • can then log in at the terminal (i.e. sit in front of a machine and type in your user name and password to log in to it). • you can also log in remotely to a machine somewhere else RAL SLAC CERN London FNAL in2p3 [email protected] 4 The command line • A user interfaces with Linux by typing commands into a shell. -

CESM2 Tutorial: Basic Modifications
CESM2 Tutorial: Basic Modifications Christine Shields August 15, 2017 CESM2 Tutorial: Basic Modifications: Review 1. We will use the CESM code located locally on Cheyenne, no need to checkout or download any input data. 2. We will run with resolution f19_g17: (atm/lnd = FV 1.9x2.5 ocn/ice=gx1v7) 3. Default scripts will automatically be configured for you using the code/script base prepared uniquely for this tutorial. 4. For On-site Tutorial ONLY: Please use compute nodes for compiling and login nodes for all other work, including submission. Please do NOT compile unless you have a compile card. To make the tutorial run smoothly for all, we need to regulate the model compiles. When you run from home, you don’t need to compile on the compute nodes. Tutorial Code and script base: /glade/p/cesm/tutorial/cesm2_0_alpha07c/ CESM2 Tutorial: Basic Modifications: Review 1. Log into Cheyenne 2. Execute create_newcase 3. Execute case.setup 4. Log onto compute node (compile_node.csH) 5. Compile model (case.build) 6. Exit compute node (type “exit”) 7. Run model (case.submit) This tutorial contains step by step instructions applicable to CESM2 (wHicH Has not been officially released yet). Documentation: Under construction! http://www.cesm.ucar.edu/models/cesm2.0/ Quick Start Guide: Under construction! http://cesm-development.github.io/cime/doc/build/html/index.Html For older releases, please see past tutorials. CESM2 Tutorial: Basic Modifications: Review: Creating a new case What is the Which Which model configuration ? Which set of components ? casename -

Xshell 6 User Guide Secure Terminal Emualtor
Xshell 6 User Guide Secure Terminal Emualtor NetSarang Computer, Inc. Copyright © 2018 NetSarang Computer, Inc. All rights reserved. Xshell Manual This software and various documents have been produced by NetSarang Computer, Inc. and are protected by the Copyright Act. Consent from the copyright holder must be obtained when duplicating, distributing or citing all or part of this software and related data. This software and manual are subject to change without prior notice for product functions improvement. Xlpd and Xftp are trademarks of NetSarang Computer, Inc. Xmanager and Xshell are registered trademarks of NetSarang Computer, Inc. Microsoft Windows is a registered trademark of Microsoft. UNIX is a registered trademark of AT&T Bell Laboratories. SSH is a registered trademark of SSH Communications Security. Secure Shell is a trademark of SSH Communications Security. This software includes software products developed through the OpenSSL Project and used in OpenSSL Toolkit. NetSarang Computer, Inc. 4701 Patrick Henry Dr. BLDG 22 Suite 137 Santa Clara, CA 95054 http://www.netsarang.com/ Contents About Xshell ............................................................................................................................................... 1 Key Functions ........................................................................................................... 1 Minimum System Requirements .................................................................................. 3 Install and Uninstall .................................................................................................. -

Penndot Form MV-409
MV-409 (3-18) www.dmv.pa.gov APPLICATION FOR CERTIFICATION OF For Department Use Only BureauP.O. Box of Motor 68697 Vehicles Harrisburg, • Vehicle PAInspection 17106-8697 Division OFFICIAL VEHICLE SAFETY INSPECTOR • PRINT OR TYPE ALL INFORMATION - MUST BE SUBMITTED TO AN APPROVED EDUCATIONAL FACILITY Applicant must be 18 years of age and have a valid operator’s license for each class of vehicle he/she intends to inspect. Applicant must also complete a lecture course at an approved educational facility, pass a written test and satisfactorily perform a complete inspection of a vehicle. upon successful completion of these courses, you will receive your certified safety inspection certification card in approximately 6 to 8 weeks from the date your class ended. The school has 35 days from the class ending date to submit the paperwork for processing. You may not begin inspecting until you receive your certification card. A APPLICANT INFORMATION Last Name First Name Middle Name Sex birth Date Driver’s License Number State r M r F Street Address City State County Zip Code Work Telephone Number Home Telephone Number Do you currently hold a valid out-of-state driver’s license? (If yes, attach a copy.) . r Yes r No *Contact PennDOT’s Vehicle Inspection Division at 717-787-2895 to establish an out-of-state mechanic record prior to completion of this class. List any restrictions on your driver’s license (if applicable): ______________________________________________________________ Do you currently hold a valid Pennsylvania driver’s license? . r Yes r No Have you held a Pennsylvania driver’s license in the past? . -
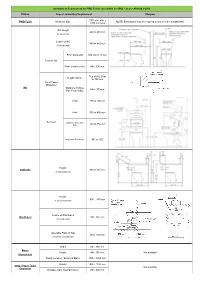
Summary of Requirement for PWD Toilets (Not Within an SOU) – As Per AS1428.1-2009
Summary of Requirement for PWD Toilets (not within an SOU) – as per AS1428.1-2009 Fixture Aspect and/or Key Requirement Diagram 1900 mm wide x PWD Toilet Minimum Size NOTE: Extra space may be required to allow for the a washbasin. 2300 mm long WC Height 460 to 480 mm (to top of seat) Centre of WC 450 to 460 mm (from side wall) From back wall 800 mm ± 10 mm Front of WC From cistern or like Min. 600 mm Top of WC Seat Height of Zone to 700 mm Toilet Paper Dispenser WC Distance of Zone Max. 300mm from Front of QC Height 150 to 200 mm Width 350 to 400 mm Backrest Distance above WC Seat 120 to 150 mm Angle from Seat Hinge 90° to 100° Height Grabrails 800 to 810 mm (to top of grab rail) Height 800 – 830 mm (to top of washbasin) Centre of Washbasin Washbasin Min. 425 mm (from side wall) Operable Parts of Tap Max. 300 mm (from front of washbasin) Width Min. 350 mm Mirror Height Min. 950 mm Not available (if provided) Fixing Location / Extent of Mirror 900 – 1850 mm Height 900 – 1100 mm Soap / Paper Towel Not available Dispenser Distance from Internal Corner Min. 500 mm Summary of Requirement for PWD Toilets (not within an SOU) – as per AS1428.1-2009 Fixture Aspect and/or Key Requirement Diagram Height 1200 – 1350 mm PWD Clothes Hook Not available Distance from Internal Corner Min. 500 mm Height 800 – 830 mm As vanity bench top Width Min. 120 mm Depth 300 – 400 mm Shelves Not available Height 900 – 1000 mm As separate fixture (if within any required Width 120 – 150 mm circulation space) Length 300 – 400 mm Width Min. -

Useful Commands in Linux and Other Tools for Quality Control
Useful commands in Linux and other tools for quality control Ignacio Aguilar INIA Uruguay 05-2018 Unix Basic Commands pwd show working directory ls list files in working directory ll as before but with more information mkdir d make a directory d cd d change to directory d Copy and moving commands To copy file cp /home/user/is . To copy file directory cp –r /home/folder . to move file aa into bb in folder test mv aa ./test/bb To delete rm yy delete the file yy rm –r xx delete the folder xx Redirections & pipe Redirection useful to read/write from file !! aa < bb program aa reads from file bb blupf90 < in aa > bb program aa write in file bb blupf90 < in > log Redirections & pipe “|” similar to redirection but instead to write to a file, passes content as input to other command tee copy standard input to standard output and save in a file echo copy stream to standard output Example: program blupf90 reads name of parameter file and writes output in terminal and in file log echo par.b90 | blupf90 | tee blup.log Other popular commands head file print first 10 lines list file page-by-page tail file print last 10 lines less file list file line-by-line or page-by-page wc –l file count lines grep text file find lines that contains text cat file1 fiel2 concatenate files sort sort file cut cuts specific columns join join lines of two files on specific columns paste paste lines of two file expand replace TAB with spaces uniq retain unique lines on a sorted file head / tail $ head pedigree.txt 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0 9 0 0 10 -

Other Useful Commands
Bioinformatics 101 – Lecture 2 Introduction to command line Alberto Riva ([email protected]), J. Lucas Boatwright ([email protected]) ICBR Bioinformatics Core Computing environments ▪ Standalone application – for local, interactive use; ▪ Command-line – local or remote, interactive use; ▪ Cluster oriented: remote, not interactive, highly parallelizable. Command-line basics ▪ Commands are typed at a prompt. The program that reads your commands and executes them is the shell. ▪ Interaction style originated in the 70s, with the first visual terminals (connections were slow…). ▪ A command consists of a program name followed by options and/or arguments. ▪ Syntax may be obscure and inconsistent (but efficient!). Command-line basics ▪ Example: to view the names of files in the current directory, use the “ls” command (short for “list”) ls plain list ls –l long format (size, permissions, etc) ls –l –t sort newest to oldest ls –l –t –r reverse sort (oldest to newest) ls –lrt options can be combined (in this case) ▪ Command names and options are case sensitive! File System ▪ Unix systems are centered on the file system. Huge tree of directories and subdirectories containing all files. ▪ Everything is a file. Unix provides a lot of commands to operate on files. ▪ File extensions are not necessary, and are not recognized by the system (but may still be useful). ▪ Please do not put spaces in filenames! Permissions ▪ Different privileges and permissions apply to different areas of the filesystem. ▪ Every file has an owner and a group. A user may belong to more than one group. ▪ Permissions specify read, write, and execute privileges for the owner, the group, everyone else. -

Install and Run External Command Line Softwares
job monitor and control top: similar to windows task manager (space to refresh, q to exit) w: who is there ps: all running processes, PID, status, type ps -ef | grep yyin bg: move current process to background fg: move current process to foreground jobs: list running and suspended processes kill: kill processes kill pid (could find out using top or ps) 1 sort, cut, uniq, join, paste, sed, grep, awk, wc, diff, comm, cat All types of bioinformatics sequence analyses are essentially text processing. Unix Shell has the above commands that are very useful for processing texts and also allows the output from one command to be passed to another command as input using pipe (“|”). less cosmicRaw.txt | cut -f2,3,4,5,8,13 | awk '$5==22' | cut -f1 | sort -u | wc This makes the processing of files using Shell very convenient and very powerful: you do not need to write output to intermediate files or load all data into the memory. For example, combining different Unix commands for text processing is like passing an item through a manufacturing pipeline when you only care about the final product 2 Hands on example 1: cosmic mutation data - Go to UCSC genome browser website: http://genome.ucsc.edu/ - On the left, find the Downloads link - Click on Human - Click on Annotation database - Ctrl+f and then search “cosmic” - On “cosmic.txt.gz” right-click -> copy link address - Go to the terminal and wget the above link (middle click or Shift+Insert to paste what you copied) - Similarly, download the “cosmicRaw.txt.gz” file - Under your home, create a folder