NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -
Chapter 13.Pptx
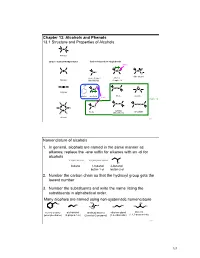
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Catalytic, Thermal, Regioselective Functionalization of Alkanes and Arenes with Borane Reagents
ACS Symposium Series 885, Activation and Functionalization of C-H Bonds, Karen I. Goldberg and Alan S. Goldman, eds. 2004. CHAP TER 8 Catalytic, Thermal, Regioselective Functionalization of Alkanes and Arenes with Borane Reagents John F. Hartwig Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107 Work in the author’s group that has led to a regioselective catalytic borylation of alkanes at the terminal position is summarized. Early findings on the photochemical, stoichiometric functionalization of arenes and alkanes and the successful extension of this work to a catalytic functionalization of alkanes under photochemical conditions is presented first. The discovery of complexes that catalyze the functionalization of alkanes to terminal alkylboronate esters is then presented, along with mechanistic studies on these system and computational work on the stoichiometric reactions of isolated metal-boryl compounds with alkanes. Parallel results on the development of catalysts and a mechanistic understanding of the borylation of arenes under mild conditions to form arylboronate esters are also presented. © 2004 American Chemical Society 136 137 1. Introduction Although alkanes are considered among the least reactive organic molecules, alkanes do react with simple elemental reagents such as halogens and oxygen.1,2) Thus, the conversion of alkanes to functionalized molecules at low temperatures with control of selectivity and at low temperatures is a focus for development of catalytic processes.(3) In particular the conversion of an alkane to a product with a functional group at the terminal position has been a longstanding goal (eq. 1). Terminal alcohols such as n-butanol and terminal amines, such as hexamethylene diamine, are major commodity chemicals(4) that are produced from reactants several steps downstream from alkane feedstocks. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

Boranes in Organic Chemistry 2. Β-Aminoalkyl- and Β-Sulfanylalkylboranes in Organic Synthesis V.M
Eurasian ChemTech Journal 4 (2002) 153-167 Boranes in Organic Chemistry 2. β-Aminoalkyl- and β-sulfanylalkylboranes in organic synthesis V.M. Dembitsky1, G.A. Tolstikov2*, M. Srebnik1 1Department of Pharmaceutical Chemistry and Natural Products, School of Pharmacy, P.O. Box 12065, The Hebrew University of Jerusalem, Jerusalem 91120, Israel 2Novosibirsk Institute of Organic Chemistry SB RAS, 9, Lavrentieva Ave., Novosibirsk, 630090, Russia Abstract Problems on using of β-aminoalkyl- and β-sulfanylalkylboranes in organic synthesis are considered in this review. The synthesis of boron containing α-aminoacids by Curtius rearrangement draws attention. The use of β-aminoalkylboranes available by enamine hydroboration are described. Examples of enamine desamination with the formation of alkenes, aminoalcohols and their transfor- mations into allylic alcohol are presented. These conversions have been carried out on steroids and nitro- gen containing heterocyclic compounds. The dihydroboration of N-vinyl-carbamate and N-vinyl-urea have been described. Examples using nitrogen and oxygen containing boron derivatives for introduction of boron functions were presented. The route to borylhydrazones by hydroboration of enehydrazones was envisaged. The possibility of trialkylamine hydroboration was shown on indole alkaloids and 11-azatricyclo- [6.2.11,802,7]2,4,6,9-undecatetraene examples. The synthesis of β-sulfanyl-alkylboranes by various routes was described. The synthesis of boronic thioaminoacids was carried out by free radical thiilation of dialkyl-vinyl- boronates. Ethoxyacetylene has been shown smoothly added 1-ethylthioboracyclopentane. Derivatives of 1,4-thiaborinane were readily obtained by divinylboronate hydroboration. Dialkylvinylboronates react with mercaptoethanol with the formation of 1,5,2-oxathioborepane derivatives. Stereochemistry of thiavinyl esters hydroboration leading to stereoisomeric β-sulfanylalkylboranes are discussed. -

Boranes: Physical & Chemical Properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-In
Boranes: Physical & chemical properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-in-Chief. International Labor Organization, Geneva. 2011. Chemical Name Colour/Form Boiling Point Melting Molecular Solubility in Relative Density Relative Vapour Inflam. Flash Auto CAS-Number (°C) Point (°C) Weight Water (water=1) Vapour Pressure/ Limits Point (°C) Ignition Density (Kpa) Point (°C) (air=1) BORON polymorphic: alpha- 2550 2300 10.81 insol Amorphous, 1.56x 580 3 -5 7440-42-8 rhombohedral form, clear 2.3 g/cm ; 10 red crystals; beta- alpha-- @ 2140 °C rhombohedral form, black; rhombohedral, - alpha-tetragonal form, 2.46 g/cm3; - black, opaque crystals with alpha-- metallic luster; amorphous tetragonal, - form, black or dark brown 2.31 g/cm3; - powder; other crystal beta-rhom- forms known bohedral, - 2.35 g/cm3 BORIC ACID, DISODIUM powder or glass-like 1575 741 201.3 2.56 g/100 g 2.367 SALT plates; white, free-flowing 1330-43-4 crystals; light grey solid BORON OXIDE rhombic crystals; 1860 450 69.6 2.77 g/100 g 1.8 1303-86-2 colourless, (amorphous); semitransparent lumps or 2.46 hard, white crystals (crystalline) BORON TRIBROMIDE colourless liquid 90 -46.0 250.57 reacts 2.6431 8.6 5.3 10294-33-4 @ 18.4 °C/4 °C @ 14 °C BORON TRICHLORIDE 12.5 -107 117.16 1.35 4.03 2.99 Pa 10294-34-5 @ 12 °C/4 @ 12.4 °C BORON TRIFLUORIDE colourless gas -99.9 -126.8 67.82 reacts 3.08g/1.57 l 2.4 10 mm Hg 7637-07-2 @ 4 °C @ -141 °C. -

Hydrogen Transmutation of Nickel in Glow Discharge
International Journal of Materials Science ISSN 0973-4589 Volume 12, Number 3 (2017), pp. 405-409 © Research India Publications http://www.ripublication.com Hydrogen Transmutation of Nickel in Glow Discharge Vladimir K. Nevolin National Research University of Electronic Technology (MIET), Moscow, Russia. Abstract Background: The possibility of the existence of subatomic hydrogen states was theoretically predicted previously. Objectives: Prove that the transmutation of elements is possible in specially prepared conditions for hydrogen. Methodology: By comparing the mass spectra of deposits on silicon substrates and target electrodes, it is shown that a change in the composition is observed in a magnetron Argon. Results: An increase in the concentration of 62 60 the nickel isotope 28 Ni and a decrease in the isotope concentration 28 Ni are shown. Conclusion: These results confirm the results obtained earlier in the heat generator Rossi, who worked more than a year, found an increase in the 62 isotope 28 Ni due to a decrease in the proportion of other isotopes. Keywords: transmutation, isotopes of nickel, glow discharge, argon, hydrogen INTRODUCION It is considered that the cold transmutation of elements (cold nuclear reactions) has been experimentally demonstrated [1]. On the basis of this phenomenon, energy generators are created in which long-term release of thermal energy in excess of expended energy is observed [2]. From many experimental studies it can be seen that hydrogen, which plays a pivotal role in the reaction zone, may be delivered through a variety of chemical compounds; for example, using lithium aluminium hydride LiAlH4. An analysis of the products of nuclear reactions suggests the possibility of many simultaneous nuclear fusion and decomposition reactions [3]. -

Boranes: Structure & Bonding
18-Aug-20 Boranes: Structure & Bonding PROF. K.K. UPADHYAY DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, BHU VARANASI - 221005 An Introduction to Boranes . For undergraduate students, boranes means only diborane which is not true. Boranes means electron deficient molecules but probably that is not true. Boranes are a class of compounds comprising of several hundreds of compounds ranging from simple to polyhedral to macro polyhedral types having intriguing structures. 1 18-Aug-20 10 11 . Boron is an element with atomic no. 5 having two isotopes 5B (20%) and 5B (80%). It occupies group XIII and is a p-block element. .Boranes are binary compounds of boron and hydrogen and are the fourth most extensive group of hydrides after the Carbon, Phosphorous and Silicon hydrides. BH 3 is the simplest of all the boranes but non-existent. B2H6 is the dimer of BH 3 and is the most primitive among the existing boranes. Boranes are not found in the nature. These are always synthesised in the laboratory. Very first synthesis was carried out in 19 th century by protolysis of metal borides. 2Mg 3B2 + 12HCl 6MgCl 2 + mixture of boranes . But neither correctly analysed nor identified. 2 18-Aug-20 . The first systematic study of boranes was performed by Alfred stock during the year 1912-1936. He used Schlenk line technique for the synthesis of boranes in a systematic way. Alfred Stock . He studied nature, stoichiometry and reactivity of boranes in a systematic way. 3 NaBH 4 + 4 BF 3→ 3 NaBF 4 + mixture of boranes BCl 3 + 3 H 2 → mixture of boranes + 3 HCl - 3- BH 4 + BX 3 → mixture of boranes + HBX (X = Cl, Br) Schlenk line 2x4 + 1x6 = 14 electrons 2x3 + 1x6 = 12 electrons .