A Mehtod for the Seperation and Gravimetric Determination of Osmium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oxidation States of Ruthenium and Osmium COMPREHENSIVE COORDINATION CHEMISTRY II
DOI: 10.1595/147106704X10801 Oxidation States of Ruthenium and Osmium COMPREHENSIVE COORDINATION CHEMISTRY II. FROM BIOLOGY TO NANOTECHNOLOGY Volume 5 TRANSITION METAL GROUPS 7 AND 8 EDITED BY E. C. CONSTABLE AND J. R. DILWORTH; EDITORS-IN-CHIEF, JON A. McCLEVERTY AND THOMAS J. MEYER, Elsevier, Amsterdam, 2003, 876 pages, ISBN 0-08-0443273 (Volume 5); ISBN 0-08-0437486 (Set), U.S.$ 5975, €6274 per Set Reviewed by C. F. J. Barnard* and S. C. Bennett Johnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading RG4 9NH, U.K.; *E-mail: [email protected] Volume 5 in the book set “Comprehensive nated by the chemistry of complexes containing Coordination Chemistry II” (CCCII) presents a the bipyridine (bpy) ligand. survey of important developments in the chemistry Many complex ligands designed to extend the of the transition metals of Groups 7 and 8: man- conjugation of the aromatic system or otherwise ganese, technetium, rhenium, iron, ruthenium (Ru) modify the electronic properties of the complex, and osmium (Os), published from 1982 to 2002. have been prepared. The complexes can be simple 2+ Volumes 6 and 9 in this 10 book set, covering mononuclear species, such as [Ru(bpy)3] , dinu- n+ work on the other platinum group metals have clear [(bpy)2Ru(µ-L)Ru(bpy)2] or polynuclear. been previously reviewed (1, 2). In Volume 5, the material for each element is organised by oxidation High Oxidation States state of the metal and also by the nature of the lig- The high oxidation states of ruthenium and ands involved, with additional sections covering osmium are areas that are generally only very light- special features of the coordination chemistry and ly covered by most chemistry reference books, applications of the complexes. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

The Periodic Table of Elements
The Periodic Table of Elements 1 2 6 Atomic Number = Number of Protons = Number of Electrons HYDROGENH HELIUMHe 1 Chemical Symbol NON-METALS 4 3 4 C 5 6 7 8 9 10 Li Be CARBON Chemical Name B C N O F Ne LITHIUM BERYLLIUM = Number of Protons + Number of Neutrons* BORON CARBON NITROGEN OXYGEN FLUORINE NEON 7 9 12 Atomic Weight 11 12 14 16 19 20 11 12 13 14 15 16 17 18 SODIUMNa MAGNESIUMMg ALUMINUMAl SILICONSi PHOSPHORUSP SULFURS CHLORINECl ARGONAr 23 24 METALS 27 28 31 32 35 40 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 POTASSIUMK CALCIUMCa SCANDIUMSc TITANIUMTi VANADIUMV CHROMIUMCr MANGANESEMn FeIRON COBALTCo NICKELNi CuCOPPER ZnZINC GALLIUMGa GERMANIUMGe ARSENICAs SELENIUMSe BROMINEBr KRYPTONKr 39 40 45 48 51 52 55 56 59 59 64 65 70 73 75 79 80 84 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 RUBIDIUMRb STRONTIUMSr YTTRIUMY ZIRCONIUMZr NIOBIUMNb MOLYBDENUMMo TECHNETIUMTc RUTHENIUMRu RHODIUMRh PALLADIUMPd AgSILVER CADMIUMCd INDIUMIn SnTIN ANTIMONYSb TELLURIUMTe IODINEI XeXENON 85 88 89 91 93 96 98 101 103 106 108 112 115 119 122 128 127 131 55 56 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 CESIUMCs BARIUMBa HAFNIUMHf TANTALUMTa TUNGSTENW RHENIUMRe OSMIUMOs IRIDIUMIr PLATINUMPt AuGOLD MERCURYHg THALLIUMTl PbLEAD BISMUTHBi POLONIUMPo ASTATINEAt RnRADON 133 137 178 181 184 186 190 192 195 197 201 204 207 209 209 210 222 87 88 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 FRANCIUMFr RADIUMRa RUTHERFORDIUMRf DUBNIUMDb SEABORGIUMSg BOHRIUMBh HASSIUMHs MEITNERIUMMt DARMSTADTIUMDs ROENTGENIUMRg COPERNICIUMCn NIHONIUMNh -

United States Patent (19) 11 Patent Number: 4,496,778 Myers Et Al
United States Patent (19) 11 Patent Number: 4,496,778 Myers et al. (45) Date of Patent: Jan. 29, 1985 (54) PROCESS FOR THE HYDROXYLATION OF 56 References Cited OLEFINS USING MOLECULAR OXYGEN, U.S. PATENT DOCUMENTS ANOSMIUM CONTAINING CATALYST, A COPPER CO-CATALYST, AND AN 2,773, 101 12/1956 Smith et al. ......................... 568/860 AROMATIC AMINE BASED PROMOTER 3,317,592 5/1967 Maclean et al. ... 568/860 3,337,635 8/1967 Norton et al. ....... 568/860 75 Inventors: Richard S. Myers, Fairlawn; Robert 4,390,739 6/1983 Michaelson et al. .... ..., 568/860 C. Michaelson, Waldwick; Richard FOREIGN PATENT DOCUMENTS G. Austin, Ridgewood, all of N.J. 32522 8/1974 Japan ................................... 568/860 73) Assignee: Exxon Research & Engineering Co., Primary Examiner-J. E. Evans Florham Park, N.J. Attorney, Agent, or Firm-Robert A. Maggio 21 Appl. No.: 538,190 57 ABSTRACT A process directed to the hydroxylation of olefins by 22 Filed: Oct. 3, 1983 reacting said olefins in the presence of oxygen, water, and a catalyst composition comprising (i) a catalytically 51 Int. Cl. ...................... C07C 29/04; CO7C 31/18; active osmium containing compound, (ii) a Co-catalyst C07C 31/22; CO7C 31/42 I comprising a copper containing compound such as 52 U.S.C. ................................. 568/860; 260/.397.2; CuBr2, and (iii) a Co-catalyst II capable of increasing 560/186; 562/587; 568/811; 568/821; 568/833; the rate and/or selectivity of the hydroxylation reac 568/838; 568/847 tion, such as pyridine is disclosed. 58 Field of Search .............. -

Precipitation of Solid Transmutation Elements in Irradiated Tungsten Alloys
Materials Transactions, Vol. 49, No. 10 (2008) pp. 2259 to 2264 #2008 The Japan Institute of Metals Precipitation of Solid Transmutation Elements in Irradiated Tungsten Alloys Takashi Tanno1;*1, Akira Hasegawa1, Mitsuhiro Fujiwara1, Jian-Chao He1;*1, Shuhei Nogami1, Manabu Satou1, Toetsu Shishido2 and Katsunori Abe1;*2 1Department of Quantum Science and Energy Engineering, Graduate School of Engineering, Tohoku University, Sendai 980-8579, Japan 2Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan Tungsten-based model alloys were fabricated to simulate compositional changes by neutron irradiation, performed in the JOYO fast test reactor. The irradiation damage range was 0.17–1.54 dpa and irradiation temperatures were 400, 500 and 750C. After irradiation, microstructural observations and electrical resistivity measurements were carried out. A number of precipitates were observed after 1.54 dpa irradiation. Rhenium and osmium were precipitated by irradiation, which suppressed the formation of dislocation loops and voids. Structures induced by irradiation were not observed so much after 0.17 dpa irradiation. Electrical resistivity measurements showed that the effects of osmium on the electrical resistivity, related to impurity solution content, were larger than that of rhenium. Measurements of electrical resistivity of ternary alloys showed that the precipitation behavior was similar to that in binary alloys. [doi:10.2320/matertrans.MAW200821] (Received April 23, 2008; Accepted August 8, 2008; Published September 18, -

The Separation and Determination of Osmium and Ruthenium
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1969 The epS aration and Determination of Osmium and Ruthenium. Harry Edward Moseley Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Moseley, Harry Edward, "The eS paration and Determination of Osmium and Ruthenium." (1969). LSU Historical Dissertations and Theses. 1559. https://digitalcommons.lsu.edu/gradschool_disstheses/1559 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. ThU dissertation has been 69-17,123 microfilmsd exactly as received MOSELEY, Harry Edward, 1929- THE SEPARATION AND DETERMINATION OF OSMIUM AND RUTHENIUM. Louisiana State University and Agricultural and Mechanical College, PhJ>., 1969 Chemistry, analytical University Microfilms, Inc., Ann Arbor, Michigan THE SEPARATION AND DETERMINATION OF OSMIUM AND RUTHENIUM A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry Harry Edward Moseley B.S., Lpuisiana State University, 1951 M.S., Louisiana State University, 1952 J anuary, 1969 ACKNOWLEDGMENTS Thanks are due to Dr. Eugene W. Berg under whose direction this work was performed, to Dr. A. D. Shendrikar for his help in the tracer studies, and to Mr. J. H. R. Streiffer for his help in writing the com puter program. -

Cellular Uptake and Toxicological Effects of Differently Sized Zinc Oxide Nanoparticles in Intestinal Cells †
toxics Article Cellular Uptake and Toxicological Effects of Differently Sized Zinc Oxide Nanoparticles in Intestinal Cells † Anna Mittag 1,* , Christian Hoera 2, Alexander Kämpfe 2 , Martin Westermann 3, Jochen Kuckelkorn 4, Thomas Schneider 1 and Michael Glei 1 1 Department of Nutritional Toxicology, Institute of Nutritional Sciences, Friedrich Schiller University Jena, Dornburger Straße 24, 07743 Jena, Germany; [email protected] (T.S.); [email protected] (M.G.) 2 German Environment Agency, Swimming Pool Water, Chemical Analytics, Heinrich-Heine-Straße 12, 08645 Bad Elster, Germany; [email protected] (C.H.); [email protected] (A.K.) 3 Electron Microscopy Centre, Friedrich Schiller University Jena, Ziegelmühlenweg 1, 07743 Jena, Germany; [email protected] 4 German Environment Agency, Toxicology of Drinking Water and Swimming Pool Water, Heinrich-Heine-Straße 12, 08645 Bad Elster, Germany; [email protected] * Correspondence: [email protected] † In respectful memory of Dr. Tamara Grummt. Abstract: Due to their beneficial properties, the use of zinc oxide nanoparticles (ZnO NP) is constantly increasing, especially in consumer-related areas, such as food packaging and food additives, which is leading to an increased oral uptake of ZnO NP. Consequently, the aim of our study was to investigate the cellular uptake of two differently sized ZnO NP (<50 nm and <100 nm; 12–1229 µmol/L) using two human intestinal cell lines (Caco-2 and LT97) and to examine the possible resulting toxic effects. ZnO NP (<50 nm and <100 nm) were internalized by both cell lines and led to intracellular changes. Citation: Mittag, A.; Hoera, C.; Kämpfe, A.; Westermann, M.; Both ZnO NP caused time- and dose-dependent cytotoxic effects, especially at concentrations of Kuckelkorn, J.; Schneider, T.; Glei, M. -

Hazardous Laboratory Chemicals Disposal Guide
Third Edition HAZARDOUS LABORATORY CHEMICALS DISPOSAL GUIDE Third Edition HAZARDOUS LABORATORY CHEMICALS DISPOSAL GUIDE Margaret-Ann Armour LEWIS PUBLISHERS A CRC Press Company Boca Raton London New York Washington, D.C. This edition published in the Taylor & Francis e-Library, 2005. To purchase your own copy of this or any of Taylor & Francis or Routledge’s collection of thousands of eBooks please go to http://www.ebookstore.tandf.co.uk/. Library of Congress Cataloging-in-Publication Data Armour, M.A. (Margaret-Ann) Hazardous laboratory chemicals disposal guide/Margaret-Ann Armour.—3rd ed. p. cm. Includes bibliographical references. ISBN 1-56670-567-3 1. Chemical laboratories—Waste disposal. 2. Hazardous substances. I. Title. QD64.A76 2003 542′.89–dc21 2002043358 This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the authors and the publisher cannot assume responsibility for the validity of all materials or for the consequences of their use. Neither this book nor any part may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage or retrieval system, without prior permission in writing from the publisher. The consent of CRC Press LLC does not extend to copying for general distribution, for promotion, for creating new works, or for resale. Specific permission must be obtained in writing from CRC Press LLC for such copying. Direct all inquiries to CRC Press LLC, 2000 N.W. -

The Two Faces of Titanium Dioxide Nanoparticles Bio-Camouflage in 3D
www.nature.com/scientificreports OPEN The two faces of titanium dioxide nanoparticles bio-camoufage in 3D bone spheroids Received: 23 October 2018 W. Souza1,2,3, S. G. Piperni3,4, P. Laviola1,3,5, A. L. Rossi4, Maria Isabel D. Rossi6, Accepted: 11 June 2019 Bráulio S. Archanjo7, P. E. Leite 1,2,8, M. H. Fernandes9,12, L. A. Rocha3,10, J. M. Granjeiro1,2,3,11 Published: xx xx xxxx & A. R. Ribeiro 2,3,5 Titanium (Ti) and its alloys are widely used in dental implants and hip-prostheses due to their excellent biocompatibility. Growing evidence support that surface degradation due to corrosion and wear processes, contribute to implant failure, since the release of metallic ions and wear particles generate local tissue reactions (peri-implant infammatory reactions). The generated ions and wear debris (particles at the micron and nanoscale) stay, in a frst moment, at the interface implant-bone. However, depending on their size, they can enter blood circulation possibly contributing to systemic reactions and toxicities. Most of the nanotoxicological studies with titanium dioxide nanoparticles (TiO2 NPs) use conventional two-dimensional cell culture monolayers to explore macrophage and monocyte activation, where limited information regarding bone cells is available. Recently three- dimensional models have been gaining prominence since they present a greater anatomical and physiological relevance. Taking this into consideration, in this work we developed a human osteoblast- like spheroid model, which closely mimics bone cell-cell interactions, providing a more realistic scenario for nanotoxicological studies. The treatment of spheroids with diferent concentrations of TiO2 NPs during 72 h did not change their viability signifcantly. -

United States Patent Office Attented Oct
3,536,479 United States Patent Office attented Oct. 27, 1970 2 a hydrogen pressure of at least about 15 pounds per 3,536,479 square inch gauge (p.S.i.g.). METHOD FOR THE PRODUCTION OF HIGH PURTY (OSMUM The concentrated osmium-containing solution can ad Alexander Ellis, Clarkson, Ontario, and Alan Manson, vantageously be prepared from a dilute osmium-contain Oakville, Ontario, Canada, assignors to The Interna 5 ing solution as described hereinafter. However, most gen tional Nicke Company, Inc., New York, N.Y., a cor erally the concentrated solution will be prepared by poration of Delaware scrubbing gases containing osmium tetroxide with a Solu No Drawing. Filled Dec. 13, 1967, Ser. No. 690,083 tion containing, by weight, about 5% to 40% sodium hy Claims priority, application Canada, Feb. 21, 1967, droxide to produce a sodium perosmate solution which 983,421 10 contains about 5 grams per liter (gp.l.) to about 100 nt. C. C22b 7/00, 11/04 g.p.l. of osmium, e.g., about 60 g.p.l. of osmium. Gen U.S. C. 75-108 29 Claims erally at this stage only minor amounts of ruthenium, i.e., less than about 0.5 g.p.l. of ruthenium, accompany the osmium and can be precipitated with only minor ABSTRACT OF THE DISCLOSURE 5 amounts of osmium occluded therein from the concen Metallic osmium is recovered from a slurry of an trated osmium-containing solution by adding a Water osmium-containing material to which sufficient hydro soluble, mild organic reducing agent such as methyl al chloric acid has been added to assure a final pH value cohol and ethyl alcohol. -

Microbial Dissolution and Stabilization of Toxic Metals and Radionuclides In
840 Experientia 46 (1990), Birkh~iuser Verlag, CH-4010 Basel/Switzerland Reviews 34 Trevors, J. T., Stratton, G. W., and Gadd, G. M., Cadmium transport, 38 Tsezos, M., and Volesky, B., The mechanism of uranium biosorption resistance and toxicity in bacteria, algae and fungi. Can. J. Microbiol. by Rhizopus arrhizus. Biotechnol. Bioeng. 24 (1982) 385-401. 32 (1986) 447-464. 39 Wainwright, M., and Grayston, S. J., Accumulation and oxidation of 35 Tsezos, M., Recovery of uranium from biological adsorbents-desorp- metal sulphides by fungi, in: Metal-Microbe Interactions, pp. 119- tion equilibrium. Biotechnol. Bioeng. 26 (1984) 973-981. 130. IRL Press, Oxford 1989. 36 Tsezos, M., Absorption by microbial biomass as a process for removal of ions from process or waste solutions, in: Immobilization of Ions by Bio-sorption, pp. 201-218. Ellis Horwood, Chichester 1986. 37 Tsezos, M., and Volesky, B., Biosorption of uranium and thorium. 0014-4754/90/080834-0751.50 + 0.20/0 Biotechnol. Bioeng. 22 (1981) 583-604. Birkhfiuser Verlag Basel, 1990 Microbial dissolution and stabilization of toxic metals and radionucfides in mixed wastes A. J. Francis Department of Applied Science, Brookhaven National Laboratory, Upton (New York 11973, USA) Summary. Microbial activity in mixed wastes can have an appreciable effect on the dissolution or precipitation of toxic metals and radionuclides. Fundamental information on microbial dissolution and stabilization (immobilization) of toxic metals and radionuclides, in particular actinides and fission products, in nuclear wastes under various microbial process conditions, e.g., aerobic, denitrifying, iron-reducing, fermentative, sulfate-reducing, and methanogenic conditions is very limited. Microbial transformations of typical waste components such as metal oxides, metal coprecipitates, naturally occurring minerals, and metal organic complexes are reviewed. -
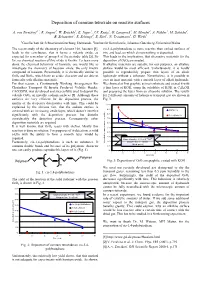
Deposition of Osmium Tetroxide on Reactive Surfaces
Deposition of osmium tetroxide on reactive surfaces A. von Zweidorf1,2, R. Angert1, W. Brüchle1, E. Jäger1, J.V. Kratz2, G. Langrock2, M. Mendel2, A. Nähler2, M. Schädel1, B. Schausten1, E. Schimpf1, E. Stiel1, N. Trautmann2, G. Wirth1 1Gesellschaft für Schwerionenforschung, Darmstadt, 2Institut für Kernchemie, Johannes Gutenberg-Universität Mainz The recent study of the chemistry of element 108, hassium [1], cis-1,4-polybutadiene is more reactive than etched surfaces of leads to the conclusion, that it forms a volatile oxide, as zinc and lead, on which almost nothing is deposited. expected for a member of group 8 of the periodic table [2]. So This leads to the implication, that alternative materials for the far, no chemical reaction of this oxide is known. To learn more deposition of OsO4 are needed. about the chemical behaviour of hassium, one would like to If alkaline materials are suitable for our purposes, an alkaline investigate the chemistry of hassium oxide, the only known surface would be most efficient. Unfortunately, it is hardly compound of hassium. Presumably, it is chemically similar to possible to reproducibly prepare thin layers of an alkali OsO4 and RuO4, which have an acidic character and are able to hydroxide without a substrate. Nevertheless, is it possible to form salts with alkaline materials. coat an inert material with a smooth layer of alkali hydroxide. For that reason, a Continuously Working Arrangement For We choosed at first graphite as inert substrate and coated it with Clusterless Transport Of In-situ Produced Volatile Oxides, a thin layer of KOH, using the solubility of KOH in C2H5OH CALLISTO, was developed and successfully used to deposit the and preparing the layer from an ethanolic solution.