Deciphering the Binding Interactions Between Acinetobacter Baumannii ACP and Β-Ketoacyl ACP Synthase III to Improve Antibiotic Targeting Using NMR Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

ATP-Citrate Lyase Has an Essential Role in Cytosolic Acetyl-Coa Production in Arabidopsis Beth Leann Fatland Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2002 ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis Beth LeAnn Fatland Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Molecular Biology Commons, and the Plant Sciences Commons Recommended Citation Fatland, Beth LeAnn, "ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis " (2002). Retrospective Theses and Dissertations. 1218. https://lib.dr.iastate.edu/rtd/1218 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis by Beth LeAnn Fatland A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Plant Physiology Program of Study Committee: Eve Syrkin Wurtele (Major Professor) James Colbert Harry Homer Basil Nikolau Martin Spalding Iowa State University Ames, Iowa 2002 UMI Number: 3158393 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -
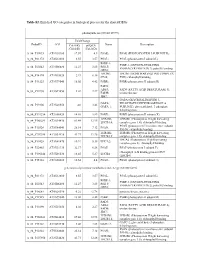
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4 -

Fatty Acid Biosynthesis
BI/CH 422/622 ANABOLISM OUTLINE: Photosynthesis Carbon Assimilation – Calvin Cycle Carbohydrate Biosynthesis in Animals Gluconeogenesis Glycogen Synthesis Pentose-Phosphate Pathway Regulation of Carbohydrate Metabolism Anaplerotic reactions Biosynthesis of Fatty Acids and Lipids Fatty Acids contrasts Diversification of fatty acids location & transport Eicosanoids Synthesis Prostaglandins and Thromboxane acetyl-CoA carboxylase Triacylglycerides fatty acid synthase ACP priming Membrane lipids 4 steps Glycerophospholipids Control of fatty acid metabolism Sphingolipids Isoprene lipids: Cholesterol ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1 ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1. Biosynthesis of fatty acids 2. Regulation of fatty acid degradation and synthesis 3. Assembly of fatty acids into triacylglycerol and phospholipids 4. Metabolism of isoprenes a. Ketone bodies and Isoprene biosynthesis b. Isoprene polymerization i. Cholesterol ii. Steroids & other molecules iii. Regulation iv. Role of cholesterol in human disease ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Degradation Synthesis Ketone body Isoprene Utilization Biosynthesis 2 Catabolism Fatty Acid Biosynthesis Anabolism • Contrast with Sugars – Lipids have have hydro-carbons not carbo-hydrates – more reduced=more energy – Long-term storage vs short-term storage – Lipids are essential for structure in ALL organisms: membrane phospholipids • Catabolism of fatty acids –produces acetyl-CoA –produces reducing -

Lipid Metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives
cancers Review Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives 1, 1, 1 2 1 Laurence Pellerin y, Lorry Carrié y , Carine Dufau , Laurence Nieto , Bruno Ségui , 1,3 1, , 1, , Thierry Levade , Joëlle Riond * z and Nathalie Andrieu-Abadie * z 1 Centre de Recherches en Cancérologie de Toulouse, Equipe Labellisée Fondation ARC, Université Fédérale de Toulouse Midi-Pyrénées, Université Toulouse III Paul-Sabatier, Inserm 1037, 2 avenue Hubert Curien, tgrCS 53717, 31037 Toulouse CEDEX 1, France; [email protected] (L.P.); [email protected] (L.C.); [email protected] (C.D.); [email protected] (B.S.); [email protected] (T.L.) 2 Institut de Pharmacologie et de Biologie Structurale, CNRS, Université Toulouse III Paul-Sabatier, UMR 5089, 205 Route de Narbonne, 31400 Toulouse, France; [email protected] 3 Laboratoire de Biochimie Métabolique, CHU Toulouse, 31059 Toulouse, France * Correspondence: [email protected] (J.R.); [email protected] (N.A.-A.); Tel.: +33-582-7416-20 (J.R.) These authors contributed equally to this work. y These authors jointly supervised this work. z Received: 15 September 2020; Accepted: 23 October 2020; Published: 27 October 2020 Simple Summary: Melanoma is a devastating skin cancer characterized by an impressive metabolic plasticity. Melanoma cells are able to adapt to the tumor microenvironment by using a variety of fuels that contribute to tumor growth and progression. In this review, the authors summarize the contribution of the lipid metabolic network in melanoma plasticity and aggressiveness, with a particular attention to specific lipid classes such as glycerophospholipids, sphingolipids, sterols and eicosanoids. -

Saturated Long-Chain Fatty Acid-Producing Bacteria Contribute
Zhao et al. Microbiome (2018) 6:107 https://doi.org/10.1186/s40168-018-0492-6 RESEARCH Open Access Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats Ling Zhao1†, Yufen Huang2†, Lin Lu1†, Wei Yang1, Tao Huang1, Zesi Lin3, Chengyuan Lin1,4, Hiuyee Kwan1, Hoi Leong Xavier Wong1, Yang Chen5, Silong Sun2, Xuefeng Xie2, Xiaodong Fang2,5, Huanming Yang6, Jian Wang6, Lixin Zhu7* and Zhaoxiang Bian1* Abstract Background: The gut microbiota is closely associated with gastrointestinal (GI) motility disorder, but the mechanism(s) by which bacteria interact with and affect host GI motility remains unclear. In this study, through using metabolomic and metagenomic analyses, an animal model of neonatal maternal separation (NMS) characterized by accelerated colonic motility and gut dysbiosis was used to investigate the mechanism underlying microbiota-driven motility dysfunction. Results: An excess of intracolonic saturated long-chain fatty acids (SLCFAs) was associated with enhanced bowel motility in NMS rats. Heptadecanoic acid (C17:0) and stearic acid (C18:0), as the most abundant odd- and even- numbered carbon SLCFAs in the colon lumen, can promote rat colonic muscle contraction and increase stool frequency. Increase of SLCFAs was positively correlated with elevated abundances of Prevotella, Lactobacillus, and Alistipes. Functional annotation found that the level of bacterial LCFA biosynthesis was highly enriched in NMS group. Essential synthetic genes Fabs were largely identified from the genera Prevotella, Lactobacillus, and Alistipes. Pseudo germ-free (GF) rats receiving fecal microbiota from NMS donors exhibited increased defecation frequency and upregulated bacterial production of intracolonic SLCFAs. Modulation of gut dysbiosis by neomycin effectively attenuated GI motility and reduced bacterial SLCFA generation in the colon lumen of NMS rats. -

Crystal Structure of Fabz-ACP Complex Reveals a Dynamic Seesaw-Like Catalytic Mechanism of Dehydratase in Fatty Acid Biosynthesis
Cell Research (2016) 26:1330-1344. ORIGINAL ARTICLE www.nature.com/cr Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis Lin Zhang1, 2, Jianfeng Xiao3, Jianrong Xu1, Tianran Fu1, Zhiwei Cao1, Liang Zhu1, 2, Hong-Zhuan Chen1, 2, Xu Shen3, Hualiang Jiang3, Liang Zhang1, 2 1Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China; 2Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai, China; 3State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China Fatty acid biosynthesis (FAS) is a vital process in cells. Fatty acids are essential for cell assembly and cellular me- tabolism. Abnormal FAS directly correlates with cell growth delay and human diseases, such as metabolic syndromes and various cancers. The FAS system utilizes an acyl carrier protein (ACP) as a transporter to stabilize and shuttle the growing fatty acid chain throughout enzymatic modules for stepwise catalysis. Studying the interactions between enzymatic modules and ACP is, therefore, critical for understanding the biological function of the FAS system. How- ever, the information remains unclear due to the high flexibility of ACP and its weak interaction with enzymatic mod- ules. We present here a 2.55 Å crystal structure of type II FAS dehydratase FabZ in complex with holo-ACP, which exhibits a highly symmetrical FabZ hexamer-ACP3 stoichiometry with each ACP binding to a FabZ dimer subunit. Further structural analysis, together with biophysical and computational results, reveals a novel dynamic seesaw-like ACP binding and catalysis mechanism for the dehydratase module in the FAS system, which is regulated by a critical gatekeeper residue (Tyr100 in FabZ) that manipulates the movements of the β-sheet layer. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

New Mechanisms That Regulate the Expression of Genes Implicated in the Process of Ketogenesis
Isabel Alexandra Pinto Carrilho do Rosário Licenciatura em Bioquímica New mechanisms that regulate the expression of genes implicated in the process of ketogenesis Dissertação para obtenção do Grau de Mestre em Biotecnologia Orientador: Prof. Dr. Pedro F. Marrero González, Prof. Titular, Facultat de Farmàcia, Universitat de Barcelona Co-orientador: Prof. Dr. Diego Haro Bautista, Prof. Catedrático, Facultat de Farmàcia, Universitat de Barcelona Presidente: Prof. Doutora Isabel Maria Godinho de Sá Nogueira Arguente: Prof. Doutor Pedro Miguel Ribeiro Viana Baptista Setembro, 2012 Isabel Alexandra Pinto Carrilho do Rosário Licenciatura em Bioquímica New mechanisms that regulate the expression of genes implicated in the process of ketogenesis Dissertação para obtenção do Grau de Mestre em Biotecnologia Orientador: Prof. Dr. Pedro F. Marrero González, Prof. Titular, Facultat de Farmàcia, Universitat de Barcelona Co-orientador: Prof. Dr. Diego Haro Bautista, Prof. Catedrático, Facultat de Farmàcia, Universitat de Barcelona Setembro, 2012 Copyright New mechanisms that regulate the expression of genes implicated in the process of ketogenesis © Isabel Alexandra Pinto Carrilho do Rosário FCT/UNL UNL A Faculdade de Ciências e Tecnologia e a Universidade Nova de Lisboa têm o direito, perpétuo e sem limites geográficos, de arquivar e publicar esta dissertação através de exemplares impressos reproduzidos em papel ou de forma digital, ou por qualquer outro meio conhecido ou que venha a ser inventado, e de a divulgar através de repositórios científicos e de admitir a sua cópia e distribuição, com objectivos educacionais ou de investigação, não comerciais, desde que seja dado crédito ao autor e editor. i ii Ninguém sabe que coisa quer. Ninguém conhece que alma tem, Nem o que é mal nem o que o bem. -

Ketogenesis Prevents Diet-Induced Fatty Liver Injury and Hyperglycemia David G
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2014 Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia David G. Cotter Washington University School of Medicine in St. Louis Baris Ercal Washington University School of Medicine in St. Louis Xiaojing Huang Washington University School of Medicine in St. Louis Jamison M. Leid Washington University School of Medicine in St. Louis Andre d'Avignon Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Cotter, David G.; Ercal, Baris; Huang, Xiaojing; Leid, Jamison M.; d'Avignon, Andre; Graham, Mark J.; Dietzen, Dennis J.; Brunt, Elizabeth M.; Patti, Gary J.; and Crawford, Peter A., ,"Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia." The Journal of Clinical Investigation.124,12. 5175-5190. (2014). https://digitalcommons.wustl.edu/open_access_pubs/3617 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors David G. Cotter, Baris Ercal, Xiaojing Huang, Jamison M. Leid, Andre d'Avignon, Mark J. Graham, Dennis J. Dietzen, Elizabeth M. Brunt, Gary J. Patti, and Peter A. Crawford This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/3617 Downloaded from http://www.jci.org on January 7, 2015. http://dx.doi.org/10.1172/JCI76388 The Journal of Clinical Investigation RESEARCH ARTICLE Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia David G.