Identification, Characterization and Evolution of Membrane-Bound Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Strategies to Increase ß-Cell Mass Expansion
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Strategies to increase -cell mass expansion Drynda, Robert Lech Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 Strategies to increase β-cell mass expansion A thesis submitted by Robert Drynda For the degree of Doctor of Philosophy from King’s College London Diabetes Research Group Division of Diabetes & Nutritional Sciences Faculty of Life Sciences & Medicine King’s College London 2017 Table of contents Table of contents ................................................................................................. -

Edinburgh Research Explorer
Edinburgh Research Explorer International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list Citation for published version: Davenport, AP, Alexander, SPH, Sharman, JL, Pawson, AJ, Benson, HE, Monaghan, AE, Liew, WC, Mpamhanga, CP, Bonner, TI, Neubig, RR, Pin, JP, Spedding, M & Harmar, AJ 2013, 'International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands', Pharmacological reviews, vol. 65, no. 3, pp. 967-86. https://doi.org/10.1124/pr.112.007179 Digital Object Identifier (DOI): 10.1124/pr.112.007179 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Pharmacological reviews Publisher Rights Statement: U.S. Government work not protected by U.S. copyright General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 1521-0081/65/3/967–986$25.00 http://dx.doi.org/10.1124/pr.112.007179 PHARMACOLOGICAL REVIEWS Pharmacol Rev 65:967–986, July 2013 U.S. -

Human Lysosomal Sulphate Transport
Human Lysosomal Sulphate Transport Martin David LEWIS B.Sc. (Hons) Thesis Submitted For the Degree Of Doctor of PhilosoPhY rn The University of Adelaide (Faculty of Medicine) May 2001 Lysosomal Diseases Research Unit and Department of P aediatrics PathologY Faculty of Medicine Department of Chemical 'Women's Women's and Children's HosPital and Children's HosPital South Australia South Australia 11 Elliot, Sømuel and Millie Table of Gontents Abstract xl1 Declaration xiv Acknowledgments XV Abbreviations xvi List of Figures xxi List of Tables xxiii 1. INTRODUGTION. """""""" 1 1.1.1 1,1.2 Membrane proteins. """"""""2 1.1.3 Types oftransporters. """"""'4 5 t.t.4 Carrier transport mechanisms. 1.1.5 1.2 Sutphate metabolism. 8 1.2.1 Phosphoadenosinephosphosulphatesynthesis"" I 1 1.2.2 The roles of sulphate within the cell' ' "" " " 1.2.2.1 A structuralrole of sulphation. ..""""' """""""""""' 11 t2 1.2.2.2 Metabolic and regulatory roles of sulphate' """""" 1.2.3 Intracellular sulphate pools...'........ """"""12 12 1.2.3.1 The origins of intracellular sulphate pools"" 1.2.3.2 Metabolism of sulphate from cysteine """""""""""" 15 1.2.3.3 Regulation of sulphate pools 1.3 The definition and function of the lysosome' """""""""""'19 1.3.1 Structure of the lysosome. ....'......... """""'20 1.3.1.1 Lysosomal hydrolysis of glycosaminoglycans"""' """"""""""""'21 25 1.3.1.1.1 Sulphatases 1.3 .2 Lysosomal biogenesis. 1.3.2.1 Targeting of lysosomal luminal proterns' 27 1V t.3.2.2 Targeting of lysosomal membrane proteins. """""""28 28 1.3.2.3 Lysosomal membrane Proteins 30 1.3.3 Lysosomal transporters 32 1.3.3.1 Proton pump (H*-ATPase) 33 1.3.3.2 Lysosomal cystine üansport............ -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners. -
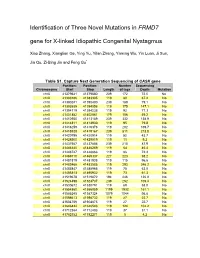
Identification of Three Novel Mutations in FRMD7 Gene for X-Linked Idiopathic Congenital Nystagmus
Identification of Three Novel Mutations in FRMD7 gene for X-linked Idiopathic Congenital Nystagmus Xiao Zhang, Xianglian Ge, Ying Yu, Yilan Zhang, Yaming Wu, Yin Luan, Ji Sun, Jia Qu, Zi-Bing Jin and Feng Gu* Table S1. Capture Next Generation Sequencing of CASK gene Position: Position: Number Sequencing Chromosome Start Stop Length of tags Depth Mutation chrX 41379641 41379880 239 172 72.0 No chrX 41383186 41383305 119 80 67.2 No chrX 41390241 41390480 239 189 79.1 No chrX 41393939 41394058 119 175 147.1 No chrX 41394119 41394238 119 92 77.3 No chrX 41401882 41402061 179 106 59.2 No chrX 41412950 41413189 239 332 138.9 No chrX 41414811 41414930 119 95 79.8 No chrX 41416259 41416378 119 202 169.7 No chrX 41418928 41419167 239 511 213.8 No chrX 41420795 41420914 119 52 43.7 No chrX 41428900 41429019 119 11 9.2 No chrX 41437567 41437806 239 210 87.9 No chrX 41446140 41446259 119 54 45.4 No chrX 41448747 41448866 119 86 72.3 No chrX 41469110 41469337 227 223 98.2 No chrX 41481819 41481938 119 115 96.6 No chrX 41483466 41483585 119 293 246.2 No chrX 41485847 41485966 119 75 63.0 No chrX 41495813 41495932 119 73 61.3 No chrX 41519678 41519872 194 246 126.8 No chrX 41524498 41524737 239 252 105.4 No chrX 41530672 41530791 119 69 58.0 No chrX 41554860 41556059 1199 1932 161.1 No chrX 41586245 41587324 1079 1044 96.8 No chrX 41598613 41598732 119 27 22.7 No chrX 41604756 41604875 119 27 22.7 No chrX 41646424 41646543 119 124 104.2 No chrX 41712364 41712483 119 37 31.1 No chrX 41782152 41782271 119 5 4.2 No Table S2. -

Supporting Information
Supporting Information Table S1. List of confirmed SLC transporters represented in Canine GeneChip. SLC family Members detected Members not detected SLC1: The high affinity glutamate and neutral amino acid SLC1A1 SLC1A2, SLC1A3, SLC1A6 transporter family SLC2: The facilitative GLUT transporter family SLC2A1, SLC2A8 SLC2A3, SLC2A9 SLC3: The heavy subunits of the heteromeric amino acid SLC3A1 transporters SLC4: The bicarbonate transporter family SLC4A11 SLC4A4, SLC4A8 SLC5: The sodium glucose cotransporter family SLC5A6 SLC5A3, SLC5A10, SLC5A12 SLC6: The sodium- and chloride- dependent SLC6A6, SLC6A12 SLCA18 neurotransmitter transporter family SLC7: The cationic amino acid transporter/glycoprotein- NR associated family SLC8: The Na+/Ca2+ exchanger family SLC8A1 SLC9: The Na+/H+ exchanger family SLC9A1, SLC9A6, SLC9A9 SLC10: The sodium bile salt cotransport family SLC10A2 SLC11: The proton coupled metal ion transporter family NR SLC12: The electroneutral cation-Cl cotransporter family SLC12A3, SLC12A6, SLC12A8 SLC13: The human Na+-sulfate/carboxylate cotransporter SLC13A2 family SLC14: The urea transporter family NR SLC15: The proton oligopeptide cotransporter family SLC15A2, SLC15A4 SLC15A1 SLC16: The monocarboxylate transporter family SLC16A13 SLC16A4 SLC17: The vesicular glutamate transporter family SLC17A3, SLC17A7 SLC18: The vesicular amine transporter family NR SLC19: The folate/thiamine transporter family NR SLC20: The type III Na+-phosphate cotransporter family NR SLC21/SLCO: The organic anion transporting family SLC21A3, SLC21A8, -

Supplementary. Table S1 a Total of Degs Detected in This Study (Gm) No
Supplementary. Table S1 A total of DEGs detected in this study (Gm) No. genename significance in annotation 1 At1g01020 D2 ARV1__expressed protein, similar to hypothetical protein DDB0188786 [Dictyostelium discoideum] (GB:EAL62332.1); contains InterPro domain Arv1-like protein (InterPro:IPR007290) 2 At1g01100 D2 60S acidic ribosomal protein P1 (RPP1A), similar to 60S ACIDIC RIBOSOMAL PROTEIN P1 GB:O23095 from (Arabidopsis thaliana) 3 At1g01120 D2, Dm KCS1__fatty acid elongase 3-ketoacyl-CoA synthase 1 (KCS1), nearly identical to GB:AAC99312 GI:4091810 from (Arabidopsis thaliana) 4 At1g01160 D1, D2, Dm GIF2__SSXT protein-related / transcription co-activator-related, similar to SYT/SSX4 fusion protein (GI:11127695) (Homo sapiens); supporting cDNA gi:21539891:gb:AY102640.1:; contains Pfam profile PF05030: SSXT protein (N-terminal region) 5 At1g01170 D2 ozone-responsive stress-related protein, putative, similar to stress-related ozone-induced protein AtOZI1 (GI:790583) (Arabidopsis thaliana); contains 1 predicted transmembrane domain; 6 At1g01240 D1, D2, Dm expressed protein 7 At1g01300 D2, Dm aspartyl protease family protein, contains Pfam domain, PF00026: eukaryotic aspartyl protease 8 At1g01320 D2 tetratricopeptide repeat (TPR)-containing protein, low similarity to SP:P46825 Kinesin light chain (KLC) {Loligo pealeii}; contains Pfam profile PF00515: TPR Domain 9 At1g01430 D2, Dm expressed protein, similar to hypothetical protein GB:CAB80917 GI:7267605 from (Arabidopsis thaliana) 10 At1g01470 D1, D2, Dm LEA14_LSR3__late embryogenesis abundant -

Br J Pharmacol
Supplementary Information Supplementary Table 1. List of polypeptide cell surface receptor and their cognate ligand genes. Supplementary Table 2. List of coding SNPs with high FST ( > 0.5) in human GPCR and their cognate ligand genes. Supplementary Table 3. List of coding SNPs with high FST ( > 0.5) in human nonGPCR receptor and ligand genes. Supplementary Table 4. List of genotyped SNPs from 44246961 to 44542055 on chromosome 17. The HapMap II dataset was analyzed using HaploView. The 101 SNPs included in LD plots of Supplementary Fig. 3 (A-C) are highlighted by a grey background. The 37 SNPs used in the haplotype analysis of Fig. 2C are indicated by red letters. SNPs that are linked with rs2291725 are indicated by bold red letters. Supplementary Table 5. Allele frequency of rs2291725 in the HGDP-CEPH populations. Frequencies of GIP103T and GIP103C alleles in each of the 52 populations from the seven geographical regions and the number of chromosomes analyzed for each population. 1 Supplementary Table 6. EC50 values for GIP receptor activation at four different time points after incubation with pooled human serum or pooled complement-preserved human serum (N=4). Supplementary Table 7. EC50 values for GIP receptor activation at three different time points after incubation with a recombinant DPP IV enzyme (N=4). Supplementary Fig. 1. Cumulative distribution function (CDF) plots for the FST of coding SNPs in human GPCRs and their cognate ligand genes (blue) and all other human genes (magenta). The FST was computed between three HapMap II populations (CEU, YRI, and ASN), and coding SNPs have been split into synonymous and nonsynonymous groups. -

Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Amy Sue Bogard University of Tennessee Health Science Center
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 12-2013 Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Amy Sue Bogard University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Medical Cell Biology Commons, and the Medical Molecular Biology Commons Recommended Citation Bogard, Amy Sue , "Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells" (2013). Theses and Dissertations (ETD). Paper 330. http://dx.doi.org/10.21007/etd.cghs.2013.0029. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Document Type Dissertation Degree Name Doctor of Philosophy (PhD) Program Biomedical Sciences Track Molecular Therapeutics and Cell Signaling Research Advisor Rennolds Ostrom, Ph.D. Committee Elizabeth Fitzpatrick, Ph.D. Edwards Park, Ph.D. Steven Tavalin, Ph.D. Christopher Waters, Ph.D. DOI 10.21007/etd.cghs.2013.0029 Comments Six month embargo expired June 2014 This dissertation is available at UTHSC Digital Commons: https://dc.uthsc.edu/dissertations/330 Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells A Dissertation Presented for The Graduate Studies Council The University of Tennessee Health Science Center In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy From The University of Tennessee By Amy Sue Bogard December 2013 Copyright © 2013 by Amy Sue Bogard. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Expression and Characterization of a Human Sodium Glucose Transporter (Hsglt1) in Pichia Pastoris
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Expression and characterization of a human sodium glucose transporter (hSGLT1) in Pichia pastoris TESI DOCTORAL Albert Suades Sala 2017 Expression and characterization of a human sodium glucose transporter (hSGLT1) in Pichia pastoris Mèmoria presentada per Albert Suades Sala per optar al grau de doctor. El treball presentat ha estat dirigit pel Dr.Josep Bartomeu Cladera Cerdà, el Dr. Joan Manyosa Ribatallada i el Dr. Alex Perálvarez Marín i realitzat en la Unitat de Biofísica del Departament de Bioquímica i de Biologia Molecular i al Centre d’Estudis en Biofísica (CEB) de la facultat de Medicina a la Universitat Autònoma de Barcelona Vist i plau dels directors de la tesi: Dr. Josep Bartomeu Cladera Dr. Joan Manyosa Ribatallada Dr. Alex Perálvarez Marín II Blessed are the forgetful, for they get the better even of their blunders Friedrich Nietzsche III Agraïments En primer lloc, m’agradaria agrair els meus directors de tesi; Joan, Pep i Alex per haver-me donat l’oportunitat i la confiança de realitzar la tesis. Al Joan, per tenir sempre preparada una broma sarcàstica sota la màniga i per animar-me de la forma més efectiva empleada a tot el món; una bona cervesa freda. -

Defining the Gene Repertoire and Spatiotemporal Expression Profiles of Adhesion G Protein-Coupled Receptors in Zebrafish Breanne L
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2015 Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish Breanne L. Harty Washington University School of Medicine in St. Louis Arunkumar Krishnan Uppsala Universitet Nicholas E. Sanchez Washington University School of Medicine in St. Louis Helgi B. Schioth Uppsala Universitet Kelly R. Monk Washington University School of Medicine in St. Louis Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Harty, Breanne L.; Krishnan, Arunkumar; Sanchez, Nicholas E.; Schioth, Helgi B.; and Monk, Kelly R., ,"Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish." BMC Genomics.16,. 62. (2015). https://digitalcommons.wustl.edu/open_access_pubs/3744 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish Harty et al. Harty et al. BMC Genomics 2015, 16: http://www.biomedcentral.com/1471-2164/16/1/ Harty et al. BMC Genomics (2015) 16:62 DOI 10.1186/s12864-015-1296-8 RESEARCH ARTICLE Open Access Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish Breanne L Harty1, Arunkumar Krishnan2, Nicholas E Sanchez1, Helgi B Schiöth2 and Kelly R Monk1,3* Abstract Background: Adhesion G protein-coupled receptors (aGPCRs) are the second largest of the five GPCR families and are essential for a wide variety of physiological processes.