Β1 and Β3 Subunits Amplify Mechanosensitivity of the Cardiac
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Towards Mutation-Specific Precision Medicine in Atypical Clinical
International Journal of Molecular Sciences Review Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes Tadashi Nakajima * , Shuntaro Tamura, Masahiko Kurabayashi and Yoshiaki Kaneko Department of Cardiovascular Medicine, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan; [email protected] (S.T.); [email protected] (M.K.); [email protected] (Y.K.) * Correspondence: [email protected]; Tel.: +81-27-220-8145; Fax: +81-27-220-8158 Abstract: Most causal genes for inherited arrhythmia syndromes (IASs) encode cardiac ion channel- related proteins. Genotype-phenotype studies and functional analyses of mutant genes, using heterol- ogous expression systems and animal models, have revealed the pathophysiology of IASs and enabled, in part, the establishment of causal gene-specific precision medicine. Additionally, the utilization of induced pluripotent stem cell (iPSC) technology have provided further insights into the patho- physiology of IASs and novel promising therapeutic strategies, especially in long QT syndrome. It is now known that there are atypical clinical phenotypes of IASs associated with specific mutations that have unique electrophysiological properties, which raises a possibility of mutation-specific precision medicine. In particular, patients with Brugada syndrome harboring an SCN5A R1632C mutation exhibit exercise-induced cardiac events, which may be caused by a marked activity-dependent loss of R1632C-Nav1.5 availability due to a marked delay of recovery from inactivation. This suggests that the use of isoproterenol should be avoided. Conversely, the efficacy of β-blocker needs to be examined. Patients harboring a KCND3 V392I mutation exhibit both cardiac (early repolarization syndrome and Citation: Nakajima, T.; Tamura, S.; paroxysmal atrial fibrillation) and cerebral (epilepsy) phenotypes, which may be associated with a Kurabayashi, M.; Kaneko, Y. -

Investigating Unexplained Deaths for Molecular Autopsies
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Investigating Unexplained Deaths for Molecular Autopsies Author(s): Yingying Tang, M.D., Ph.D, DABMG Document Number: 255135 Date Received: August 2020 Award Number: 2011-DN-BX-K535 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Technical Report NIJ FY 11 Basic Science Research to Support Forensic Science 2011-DN-BX-K535 Investigating Unexplained Deaths through Molecular Autopsies May 28, 2017 Yingying Tang, MD, PhD, DABMG Principal Investigator Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] Page 1 of 41 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Sudden Unexplained Death (SUD) is natural death in a previously healthy individual whose cause remains undetermined after scene investigation, complete autopsy, and medical record review. SUD affects children and adults, devastating families, challenging medical examiners, and is a focus of research for cardiologists, neurologists, clinical geneticists, and scientists. -

Mapping Protein Interactions of Sodium Channel Nav1.7 Using 2 Epitope-Tagged Gene Targeted Mice
bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Mapping protein interactions of sodium channel NaV1.7 using 2 epitope-tagged gene targeted mice 3 Alexandros H. Kanellopoulos1†, Jennifer Koenig1, Honglei Huang2, Martina Pyrski3, 4 Queensta Millet1, Stephane Lolignier1, Toru Morohashi1, Samuel J. Gossage1, Maude 5 Jay1, John Linley1, Georgios Baskozos4, Benedikt Kessler2, James J. Cox1, Frank 6 Zufall3, John N. Wood1* and Jing Zhao1†** 7 1Molecular Nociception Group, WIBR, University College London, Gower Street, 8 London WC1E 6BT, UK. 9 2Mass Spectrometry Laboratory, Target Discovery Institute, University of Oxford, The 10 Old Road Campus, Oxford, OX3 7FZ, UK. 11 3Center for Integrative Physiology and Molecular Medicine, Saarland University, 12 Kirrbergerstrasse, Bldg. 48, 66421 Homburg, Germany. 13 4Division of Bioscience, University College London, Gower Street, London WC1E 6BT, UK.14 15 †These authors contributed equally to directing this work. 16 *Correspondence author. Tel: +44 207 6796 954; E-mail: [email protected] 17 **Corresponding author: Tel: +44 207 6790 959; E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 18 Abstract 19 The voltage-gated sodium channel NaV1.7 plays a critical role in pain pathways. 20 Besides action potential propagation, NaV1.7 regulates neurotransmitter release, 21 integrates depolarizing inputs over long periods and regulates transcription. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
Inheritable Arrhytmia Syndromes
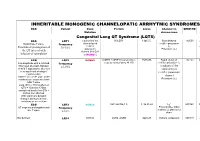
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Human Neuronal Changes in Brain Edema and Increased Intracranial
Faragó et al. Acta Neuropathologica Communications (2016) 4:78 DOI 10.1186/s40478-016-0356-x RESEARCH Open Access Human neuronal changes in brain edema and increased intracranial pressure Nóra Faragó1,2,3, Ágnes Katalin Kocsis1, Csilla Braskó1, Sándor Lovas1, Márton Rózsa1, Judith Baka1, Balázs Kovács1, Katalin Mikite1, Viktor Szemenyei1, Gábor Molnár1, Attila Ozsvár1, Gáspár Oláh1, Ildikó Piszár1, Ágnes Zvara2, Attila Patócs4, Pál Barzó5, László G. Puskás2,3 and Gábor Tamás1* Abstract Functional and molecular changes associated with pathophysiological conditions are relatively easily detected based on tissue samples collected from patients. Population specific cellular responses to disease might remain undiscovered in samples taken from organs formed by a multitude of cell types. This is particularly apparent in the human cerebral cortex composed of a yet undefined number of neuron types with a potentially different involvement in disease processes. We combined cellular electrophysiology, anatomy and single cell digital PCR in human neurons identified in situ for the first time to assess mRNA expression and corresponding functional changes in response to edema and increased intracranial pressure. In single pyramidal cells, mRNA copy numbers of AQP1, AQP3, HMOX1, KCNN4, SCN3B and SOD2 increased, while CACNA1B, CRH decreased in edema. In addition, single pyramidal cells increased the copy number of AQP1, HTR5A and KCNS1 mRNAs in response to increased intracranial pressure. In contrast to pyramidal cells, AQP1, HMOX1and KCNN4 remained unchanged in single cell digital PCR performed on fast spiking cells in edema. Corroborating single cell digital PCR results, pharmacological and immunohistochemical results also suggested the presence of KCNN4 encoding the α-subunit of KCa3.1 channels in edema on pyramidal cells, but not on interneurons. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Molecular Specializations of Deep Cortical Layer Analogs in Songbirds Alexander A
www.nature.com/scientificreports OPEN Molecular specializations of deep cortical layer analogs in songbirds Alexander A. Nevue, Peter V. Lovell, Morgan Wirthlin & Claudio V. Mello* How the evolution of complex behavioral traits is associated with the emergence of novel brain pathways is largely unknown. Songbirds, like humans, learn vocalizations via tutor imitation and possess a specialized brain circuitry to support this behavior. In a comprehensive in situ hybridization efort, we show that the zebra fnch vocal robust nucleus of the arcopallium (RA) shares numerous markers (e.g. SNCA, PVALB) with the adjacent dorsal intermediate arcopallium (AId), an avian analog of mammalian deep cortical layers with involvement in motor function. We also identify markers truly unique to RA and thus likely linked to modulation of vocal motor function (e.g. KCNC1, GABRE), including a subset of the known shared markers between RA and human laryngeal motor cortex (e.g. SLIT1, RTN4R, LINGO1, PLXNC1). The data provide novel insights into molecular features unique to vocal learning circuits, and lend support for the motor theory for vocal learning origin. An in-depth understanding of how the brain controls learned behaviors and how these behaviors arise in specifc animal lineages requires detailed knowledge of the molecular organization of the underlying circuits. Songbirds ofer an excellent model for investigating these questions. Teir vocal circuitry has been extensively studied, and consists of interconnected pallial, basal ganglia, and thalamic components that control the production and acquisition of learned vocalizations1. As is typical of birds, the pallial (cortical-like) areas consist of discrete nuclei, in contrast to the layered cortex of mammals2–4. -

Graded Co-Expression of Ion Channel, Neurofilament, and Synaptic Genes in Fast- Spiking Vestibular Nucleus Neurons
Research Articles: Cellular/Molecular Graded co-expression of ion channel, neurofilament, and synaptic genes in fast- spiking vestibular nucleus neurons https://doi.org/10.1523/JNEUROSCI.1500-19.2019 Cite as: J. Neurosci 2019; 10.1523/JNEUROSCI.1500-19.2019 Received: 26 June 2019 Revised: 11 October 2019 Accepted: 25 October 2019 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2019 the authors 1 Graded co-expression of ion channel, neurofilament, and synaptic genes in fast-spiking 2 vestibular nucleus neurons 3 4 Abbreviated title: A fast-spiking gene module 5 6 Takashi Kodama1, 2, 3, Aryn Gittis, 3, 4, 5, Minyoung Shin2, Keith Kelleher2, 3, Kristine Kolkman3, 4, 7 Lauren McElvain3, 4, Minh Lam1, and Sascha du Lac1, 2, 3 8 9 1 Johns Hopkins University School of Medicine, Baltimore MD, 21205 10 2 Howard Hughes Medical Institute, La Jolla, CA, 92037 11 3 Salk Institute for Biological Studies, La Jolla, CA, 92037 12 4 Neurosciences Graduate Program, University of California San Diego, La Jolla, CA, 92037 13 5 Carnegie Mellon University, Pittsburgh, PA, 15213 14 15 Corresponding Authors: 16 Takashi Kodama ([email protected]) 17 Sascha du Lac ([email protected]) 18 Department of Otolaryngology-Head and Neck Surgery 19 The Johns Hopkins University School of Medicine 20 Ross Research Building 420, 720 Rutland Avenue, Baltimore, Maryland, 21205 21 22 23 Conflict of Interest 24 The authors declare no competing financial interests. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias
Acta Cardiol Sin 2019;35:99-110 Review Article doi: 10.6515/ACS.201903_35(2).20181028A Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias Amir Dehghani-Samani,1 Samin Madreseh-Ghahfarokhi2 and Azam Dehghani-Samani3 Background: Arrhythmias as important malfunctions of heart are known as abnormal rhythm of heart. Several causes can make arrhythmias and most of them are related to generation and/or conduction of action potential in heart. Action potential in myocytes results from the sequential opening and closing of ion channel proteins that span the plasma membrane of individual myocytes. Action potential’s conduction through the heart is depended on electrical coupling between myocytes, which is mediated by gap junctions. Generation and conduction of action potentials are related to perfect action of ionic channels in heart. Objectives: This novel review comprehensively addressed the ionic mechanisms of the arrhythmogenic mutations in cardiac voltage-gated ionic channels including: CACNA1C, CACNA1D, KCNA5, KCND2, KCND3, KCNE1, KCNE2, KCNE5, KCNH2, KCNJ2, KCNJ5, KCNQ1, SCN4A, SCN5A, SCN1B, SCN2B, SCN3B and SCN4B. Methods: Current study, for the first time, review and discuses about relation between cardiac arrhythmias and whole of important voltage gated ionic channels from different families, altogether and at the same time. Results: This review clears that mutations in voltage-gated ionic channels play important roles in generation of severe cardiac arrhythmias, and among them it is looked that mutations in voltage-gated potassium channels are more important. Conclusions: Most of induced arrhythmias due to voltage-gated ionic channels mutations result in action potentials prolongation and long QT syndromes.