Mechanisms of Epileptogenesis Due to Mutations of Voltage-Gated Sodium Channel SCN1B By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Towards Mutation-Specific Precision Medicine in Atypical Clinical
International Journal of Molecular Sciences Review Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes Tadashi Nakajima * , Shuntaro Tamura, Masahiko Kurabayashi and Yoshiaki Kaneko Department of Cardiovascular Medicine, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan; [email protected] (S.T.); [email protected] (M.K.); [email protected] (Y.K.) * Correspondence: [email protected]; Tel.: +81-27-220-8145; Fax: +81-27-220-8158 Abstract: Most causal genes for inherited arrhythmia syndromes (IASs) encode cardiac ion channel- related proteins. Genotype-phenotype studies and functional analyses of mutant genes, using heterol- ogous expression systems and animal models, have revealed the pathophysiology of IASs and enabled, in part, the establishment of causal gene-specific precision medicine. Additionally, the utilization of induced pluripotent stem cell (iPSC) technology have provided further insights into the patho- physiology of IASs and novel promising therapeutic strategies, especially in long QT syndrome. It is now known that there are atypical clinical phenotypes of IASs associated with specific mutations that have unique electrophysiological properties, which raises a possibility of mutation-specific precision medicine. In particular, patients with Brugada syndrome harboring an SCN5A R1632C mutation exhibit exercise-induced cardiac events, which may be caused by a marked activity-dependent loss of R1632C-Nav1.5 availability due to a marked delay of recovery from inactivation. This suggests that the use of isoproterenol should be avoided. Conversely, the efficacy of β-blocker needs to be examined. Patients harboring a KCND3 V392I mutation exhibit both cardiac (early repolarization syndrome and Citation: Nakajima, T.; Tamura, S.; paroxysmal atrial fibrillation) and cerebral (epilepsy) phenotypes, which may be associated with a Kurabayashi, M.; Kaneko, Y. -

Investigating Unexplained Deaths for Molecular Autopsies
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Investigating Unexplained Deaths for Molecular Autopsies Author(s): Yingying Tang, M.D., Ph.D, DABMG Document Number: 255135 Date Received: August 2020 Award Number: 2011-DN-BX-K535 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Technical Report NIJ FY 11 Basic Science Research to Support Forensic Science 2011-DN-BX-K535 Investigating Unexplained Deaths through Molecular Autopsies May 28, 2017 Yingying Tang, MD, PhD, DABMG Principal Investigator Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] Page 1 of 41 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Sudden Unexplained Death (SUD) is natural death in a previously healthy individual whose cause remains undetermined after scene investigation, complete autopsy, and medical record review. SUD affects children and adults, devastating families, challenging medical examiners, and is a focus of research for cardiologists, neurologists, clinical geneticists, and scientists. -

Mapping Protein Interactions of Sodium Channel Nav1.7 Using 2 Epitope-Tagged Gene Targeted Mice
bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Mapping protein interactions of sodium channel NaV1.7 using 2 epitope-tagged gene targeted mice 3 Alexandros H. Kanellopoulos1†, Jennifer Koenig1, Honglei Huang2, Martina Pyrski3, 4 Queensta Millet1, Stephane Lolignier1, Toru Morohashi1, Samuel J. Gossage1, Maude 5 Jay1, John Linley1, Georgios Baskozos4, Benedikt Kessler2, James J. Cox1, Frank 6 Zufall3, John N. Wood1* and Jing Zhao1†** 7 1Molecular Nociception Group, WIBR, University College London, Gower Street, 8 London WC1E 6BT, UK. 9 2Mass Spectrometry Laboratory, Target Discovery Institute, University of Oxford, The 10 Old Road Campus, Oxford, OX3 7FZ, UK. 11 3Center for Integrative Physiology and Molecular Medicine, Saarland University, 12 Kirrbergerstrasse, Bldg. 48, 66421 Homburg, Germany. 13 4Division of Bioscience, University College London, Gower Street, London WC1E 6BT, UK.14 15 †These authors contributed equally to directing this work. 16 *Correspondence author. Tel: +44 207 6796 954; E-mail: [email protected] 17 **Corresponding author: Tel: +44 207 6790 959; E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/118497; this version posted March 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 18 Abstract 19 The voltage-gated sodium channel NaV1.7 plays a critical role in pain pathways. 20 Besides action potential propagation, NaV1.7 regulates neurotransmitter release, 21 integrates depolarizing inputs over long periods and regulates transcription. -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Inhibition of ROMK Channels by Low Extracellular K and Oxidative Stress
Am J Physiol Renal Physiol 305: F208–F215, 2013. First published May 15, 2013; doi:10.1152/ajprenal.00185.2013. Inhibition of ROMK channels by low extracellular Kϩ and oxidative stress Gustavo Frindt,1 Hui Li,2 Henry Sackin,2 and Lawrence G. Palmer1 1Department of Physiology and Biophysics, Weill-Cornell Medical College, New York, New York; and 2Department of Physiology and Biophysics, The Chicago Medical School, Rosalind Franklin University, North Chicago, Illinois Submitted 2 April 2013; accepted in final form 8 May 2013 Frindt G, Li H, Sackin H, Palmer LG. Inhibition of ROMK may be essential for preventing Kϩ secretion and minimizing channels by low extracellular Kϩ and oxidative stress. Am J Physiol K losses. Renal Physiol 305: F208–F215, 2013. First published May 15, 2013; Measurements of ROMK activity in heterologous expression doi:10.1152/ajprenal.00185.2013.—We tested the hypothesis that low systems indicate that the channels are sensitive to changes in luminal Kϩ inhibits the activity of ROMK channels in the rat cortical ϩ ϩ ϩ the extracellular K concentration ([K ]o); decreases in [K ]o collecting duct. Whole-cell voltage-clamp measurements of the com- downregulate the channels (7, 28, 29, 31). One aspect of this Downloaded from ponent of outward Kϩ current inhibited by the bee toxin Tertiapin-Q ϩ response is a shift in the dependence of channel activity on (ISK) showed that reducing the bath concentration ([K ]o)to1mM intracellular pH, with low [Kϩ] moving the titration curve for resulted in a decline of current over 2 min compared with that o ϩ inhibition of the channels toward a higher, more physiological observed at 10 mM [K ]o. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
Inheritable Arrhytmia Syndromes
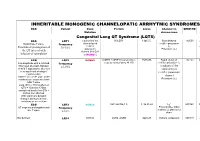
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Human Neuronal Changes in Brain Edema and Increased Intracranial
Faragó et al. Acta Neuropathologica Communications (2016) 4:78 DOI 10.1186/s40478-016-0356-x RESEARCH Open Access Human neuronal changes in brain edema and increased intracranial pressure Nóra Faragó1,2,3, Ágnes Katalin Kocsis1, Csilla Braskó1, Sándor Lovas1, Márton Rózsa1, Judith Baka1, Balázs Kovács1, Katalin Mikite1, Viktor Szemenyei1, Gábor Molnár1, Attila Ozsvár1, Gáspár Oláh1, Ildikó Piszár1, Ágnes Zvara2, Attila Patócs4, Pál Barzó5, László G. Puskás2,3 and Gábor Tamás1* Abstract Functional and molecular changes associated with pathophysiological conditions are relatively easily detected based on tissue samples collected from patients. Population specific cellular responses to disease might remain undiscovered in samples taken from organs formed by a multitude of cell types. This is particularly apparent in the human cerebral cortex composed of a yet undefined number of neuron types with a potentially different involvement in disease processes. We combined cellular electrophysiology, anatomy and single cell digital PCR in human neurons identified in situ for the first time to assess mRNA expression and corresponding functional changes in response to edema and increased intracranial pressure. In single pyramidal cells, mRNA copy numbers of AQP1, AQP3, HMOX1, KCNN4, SCN3B and SOD2 increased, while CACNA1B, CRH decreased in edema. In addition, single pyramidal cells increased the copy number of AQP1, HTR5A and KCNS1 mRNAs in response to increased intracranial pressure. In contrast to pyramidal cells, AQP1, HMOX1and KCNN4 remained unchanged in single cell digital PCR performed on fast spiking cells in edema. Corroborating single cell digital PCR results, pharmacological and immunohistochemical results also suggested the presence of KCNN4 encoding the α-subunit of KCa3.1 channels in edema on pyramidal cells, but not on interneurons. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -

Ion Channels Accelerated D1scovery
Quality Antibodies · Quality Results $-GeneTex Your Expertise Our Antibod1es Ion Channels Accelerated D1scovery --------- www.genetex.com Potassium (K+ ) Sodium (Na+ ) Calcium (Ca2+ ) Ion channels are pore-forming, usually multimeric plasma membrane proteins that can open and close in response to chemical, temperature, or mechanical signals. An open channel allows specific ions to rapid ly traverse the transmembrane passageway a long an electrochemical gradient, generating an electrical signal that is propagated along excitable cells. Ion channels are of immense importance in clinical medicine as they are linked to a broad array of disorders and are targets of an ever-expanding, and commonly prescribed, armamentarium of pharmacologic agents. Nevertheless, there remains a great void in our understanding of ion channel biology, which limits our ability to develop more effective andmo re specific drugs. GeneTex offers an outstanding selection of antibodies to support ion channel research.Please see the highlighted antibodies in this flyer or review our complete list of related products on the GeneTex website. Highlighted products HCNl antibody (GTX131334} DPP6 antibody (GTX133338} IP3 Receptor I antibody (GTX133104} (...___ ___w _w _w_ ._G_e_n_e_T_e_x_._c_o_m_ _____,) ` a n a 匱 。 'ot I: . ·""·,, " . 鼴 丶•' · ,' , `> ,,, 丶 B -- ,:: ·-- o .i :' T. 、 冨 ,. \\.◄ .•}' r<• .. , -·•Jt.<_,,,, . ◄ · ◄ / .Jj -~~ 盆;i . '. ., CACNB4 (GTX100202) VGluTl antibody (GTX133148) P2X7一 antibody (GTX104288) Cavl.2 antibody (GTX54754) Cav2.1 antibody (GTX54753)