The Cardiac-Specific N-Terminal Region of Troponin I Positions the Regulatory Domain of Troponin C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Families and the Structural Relatedness Among Globular Proteins
Protein Science (1993), 2, 884-899. Cambridge University Press. Printed in the USA. Copyright 0 1993 The Protein Society -~~ ~~~~ ~ Families and the structural relatedness among globular proteins DAVID P. YEE AND KEN A. DILL Department of Pharmaceutical Chemistry, University of California, San Francisco, California94143-1204 (RECEIVEDJanuary 6, 1993; REVISEDMANUSCRIPT RECEIVED February 18, 1993) Abstract Protein structures come in families. Are families “closely knit” or “loosely knit” entities? We describe a mea- sure of relatedness among polymer conformations. Based on weighted distance maps, this measure differs from existing measures mainly in two respects: (1) it is computationally fast, and (2) it can compare any two proteins, regardless of their relative chain lengths or degree of similarity. It does not require finding relative alignments. The measure is used here to determine the dissimilarities between all 12,403 possible pairs of 158 diverse protein structures from the Brookhaven Protein Data Bank (PDB). Combined with minimal spanning trees and hier- archical clustering methods,this measure is used to define structural families. It is also useful for rapidly searching a dataset of protein structures for specific substructural motifs.By using an analogy to distributions of Euclid- ean distances, we find that protein families are not tightly knit entities. Keywords: protein family; relatedness; structural comparison; substructure searches Pioneering work over the past 20 years has shown that positions after superposition. RMS is a useful distance proteins fall into families of related structures (Levitt & metric for comparingstructures that arenearly identical: Chothia, 1976; Richardson, 1981; Richardson & Richard- for example, when refining or comparing structures ob- son, 1989; Chothia & Finkelstein, 1990). -

CCN3 and Calcium Signaling Alain Lombet1, Nathalie Planque2, Anne-Marie Bleau2, Chang Long Li2 and Bernard Perbal*2
Cell Communication and Signaling BioMed Central Review Open Access CCN3 and calcium signaling Alain Lombet1, Nathalie Planque2, Anne-Marie Bleau2, Chang Long Li2 and Bernard Perbal*2 Address: 1CNRS UMR 8078, Hôpital Marie Lannelongue, 133, Avenue de la Résistance 92350 Le PLESSIS-ROBINSON, France and 2Laboratoire d'Oncologie Virale et Moléculaire, Tour 54, Case 7048, Université Paris 7-D.Diderot, 2 Place Jussieu 75005 PARIS, France Email: Alain Lombet - [email protected]; Nathalie Planque - [email protected]; Anne-Marie Bleau - [email protected]; Chang Long Li - [email protected]; Bernard Perbal* - [email protected] * Corresponding author Published: 15 August 2003 Received: 26 June 2003 Accepted: 15 August 2003 Cell Communication and Signaling 2003, 1:1 This article is available from: http://www.biosignaling.com/content/1/1/1 © 2003 Lombet et al; licensee BioMed Central Ltd. This is an Open Access article: verbatim copying and redistribution of this article are permitted in all media for any purpose, provided this notice is preserved along with the article's original URL. Abstract The CCN family of genes consists presently of six members in human (CCN1-6) also known as Cyr61 (Cystein rich 61), CTGF (Connective Tissue Growth Factor), NOV (Nephroblastoma Overexpressed gene), WISP-1, 2 and 3 (Wnt-1 Induced Secreted Proteins). Results obtained over the past decade have indicated that CCN proteins are matricellular proteins, which are involved in the regulation of various cellular functions, such as proliferation, differentiation, survival, adhesion and migration. The CCN proteins have recently emerged as regulatory factors involved in both internal and external cell signaling. -

Calmodulin Binding Proteins and Alzheimer's Disease
International Journal of Molecular Sciences Review Calmodulin Binding Proteins and Alzheimer’s Disease: Biomarkers, Regulatory Enzymes and Receptors That Are Regulated by Calmodulin Danton H. O’Day 1,2 1 Cell and Systems Biology, University of Toronto, Toronto, ON M5S 3G5, Canada; [email protected] 2 Department of Biology, University of Toronto Mississauga, Mississauga, ON L5L 1C6, Canada Received: 18 September 2020; Accepted: 3 October 2020; Published: 5 October 2020 Abstract: The integral role of calmodulin in the amyloid pathway and neurofibrillary tangle formation in Alzheimer’s disease was first established leading to the “Calmodulin Hypothesis”. Continued research has extended our insight into the central function of the small calcium sensor and effector calmodulin and its target proteins in a multitude of other events associated with the onset and progression of this devastating neurodegenerative disease. Calmodulin’s involvement in the contrasting roles of calcium/CaM-dependent kinase II (CaMKII) and calcineurin (CaN) in long term potentiation and depression, respectively, and memory impairment and neurodegeneration are updated. The functions of the proposed neuronal biomarker neurogranin, a calmodulin binding protein also involved in long term potentiation and depression, is detailed. In addition, new discoveries into calmodulin’s role in regulating glutamate receptors (mGluR, NMDAR) are overviewed. The interplay between calmodulin and amyloid beta in the regulation of PMCA and ryanodine receptors are prime examples of how the buildup of classic biomarkers can underly the signs and symptoms of Alzheimer’s. The role of calmodulin in the function of stromal interaction molecule 2 (STIM2) and adenosine A2A receptor, two other proteins linked to neurodegenerative events, is discussed. -

New Approach for Untangling the Role of Uncommon Calcium-Binding Proteins in the Central Nervous System
brain sciences Review New Approach for Untangling the Role of Uncommon Calcium-Binding Proteins in the Central Nervous System Krisztina Kelemen * and Tibor Szilágyi Department of Physiology, Doctoral School, Faculty of Medicine, George Emil Palade University of Medicine, Pharmacy, Science, and Technology of Targu Mures, 540142 Târgu Mures, , Romania; [email protected] * Correspondence: [email protected]; Tel.: +40-746-248064 Abstract: Although Ca2+ ion plays an essential role in cellular physiology, calcium-binding proteins (CaBPs) were long used for mainly as immunohistochemical markers of specific cell types in different regions of the central nervous system. They are a heterogeneous and wide-ranging group of proteins. Their function was studied intensively in the last two decades and a tremendous amount of informa- tion was gathered about them. Girard et al. compiled a comprehensive list of the gene-expression profiles of the entire EF-hand gene superfamily in the murine brain. We selected from this database those CaBPs which are related to information processing and/or neuronal signalling, have a Ca2+- buffer activity, Ca2+-sensor activity, modulator of Ca2+-channel activity, or a yet unknown function. In this way we created a gene function-based selection of the CaBPs. We cross-referenced these findings with publicly available, high-quality RNA-sequencing and in situ hybridization databases (Human Protein Atlas (HPA), Brain RNA-seq database and Allen Brain Atlas integrated into the HPA) and created gene expression heat maps of the regional and cell type-specific expression levels of the selected CaBPs. This represents a useful tool to predict and investigate different expression patterns and functions of the less-known CaBPs of the central nervous system. -

Cytoskeletal Remodeling in Cancer
biology Review Cytoskeletal Remodeling in Cancer Jaya Aseervatham Department of Ophthalmology, University of Texas Health Science Center at Houston, Houston, TX 77054, USA; [email protected]; Tel.: +146-9767-0166 Received: 15 October 2020; Accepted: 4 November 2020; Published: 7 November 2020 Simple Summary: Cell migration is an essential process from embryogenesis to cell death. This is tightly regulated by numerous proteins that help in proper functioning of the cell. In diseases like cancer, this process is deregulated and helps in the dissemination of tumor cells from the primary site to secondary sites initiating the process of metastasis. For metastasis to be efficient, cytoskeletal components like actin, myosin, and intermediate filaments and their associated proteins should co-ordinate in an orderly fashion leading to the formation of many cellular protrusions-like lamellipodia and filopodia and invadopodia. Knowledge of this process is the key to control metastasis of cancer cells that leads to death in 90% of the patients. The focus of this review is giving an overall understanding of these process, concentrating on the changes in protein association and regulation and how the tumor cells use it to their advantage. Since the expression of cytoskeletal proteins can be directly related to the degree of malignancy, knowledge about these proteins will provide powerful tools to improve both cancer prognosis and treatment. Abstract: Successful metastasis depends on cell invasion, migration, host immune escape, extravasation, and angiogenesis. The process of cell invasion and migration relies on the dynamic changes taking place in the cytoskeletal components; actin, tubulin and intermediate filaments. This is possible due to the plasticity of the cytoskeleton and coordinated action of all the three, is crucial for the process of metastasis from the primary site. -
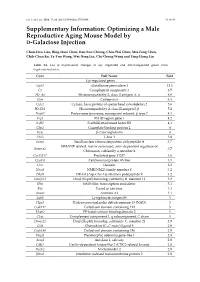
Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection
Int. J. Mol. Sci. 2016, 17, 98; doi:10.3390/ijms17010098 S1 of S4 Supplementary Information: Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection Chun-Hou Liao, Bing-Huei Chen, Han-Sun Chiang, Chiu-Wei Chen, Mei-Feng Chen, Chih-Chun Ke, Ya-Yun Wang, Wei-Ning Lin, Chi-Chung Wang and Ying-Hung Lin Table S1. List of expressional changes of up- regulated and down-regulated genes from D-gal-injected mice. Gene Full Name Fold Up-regulated genes Gpx3 Glutathione peroxidase 3 13.1 C3 Complement component 3 8.5 H2-Aa Histocompatibility 2, class II antigen A, α 6.9 Ctss Cathepsin S 6.3 Cylc2 Cylicin, basic protein of sperm head cytoskeleton 2 5.6 H2-Eb1 Histocompatibility 2, class II antigen E β 5.2 Psmb7 Proteasome (prosome, macropain) subunit, β type 7 4.3 Frg1 FSHD region gene 1 4.2 Safb2 Scaffold attachment factor B2 4.1 Gbp2 Guanylate binding protein 2 4 B2m β-2 microglobulin 4 Tbx3 T-box 3 3.8 Snrpa Small nuclear ribonucleoprotein polypeptide A 3.7 SWI/SNF related, matrix associated, actin dependent regulator of Smarca2 3.7 Chromatin, subfamily a, member 2 Gm13237 Predicted gene 13237 3.6 Cep85l Centrosomal protein 85-like 3.5 Dcn Decorin 3.2 Nme8 NME/NM23 family member 8 3.2 Dhx9 DEAH (Asp-Glu-Ala-His) box polypeptide 9 3.2 Dnajb11 DnaJ (Hsp40) homolog, subfamily B, member 11 3.2 Sltm SAFB-like, transcription modulator 3.1 Fus Fused in sarcoma 3.1 Anxa1 Annexin A1 3.1 Ly86 Lymphocyte antigen 86 3 Hpgd Hydroxyprostaglandin dehydrogenase 15 (NAD) 3 Ccdc112 Coiled-coil domain containing 112 3 Efcab2 EF-hand calcium binding domain 2 3 C1qc Complement component 1, q subcomponent, C chain 3 Dnajc21 DnaJ (Hsp40) homolog, subfamily C, member 21 2.9 Ccl8 Chemokine (C–C motif) ligand 8 2.9 Ccdc186 Coiled-coil domain containing 186 2.9 Plagl1 Pleomorphic adenoma gene-like 1 2.8 Amy1 Amylase 1, salivary 2.8 Cdkl2 Cyclin-dependent kinase-like 2 (CDC2-related kinase) 2.8 Nucb2 Nucleobindin 2 2.8 Ifitm2 Interferon induced transmembrane protein 2 2.8 Int. -

Anthopleura Elegantissima ⁎ Laura L
Comparative Biochemistry and Physiology, Part B 146 (2007) 551–559 www.elsevier.com/locate/cbpb Characterization of a novel EF-hand homologue, CnidEF, in the sea anemone Anthopleura elegantissima ⁎ Laura L. Hauck , Wendy S. Phillips, Virginia M. Weis Department of Zoology, 3029 Cordley Hall, Oregon State University, Corvallis, Oregon 97331, USA Received 25 May 2006; received in revised form 1 December 2006; accepted 3 December 2006 Available online 12 December 2006 Abstract The superfamily of EF-hand proteins is comprised of a large and diverse group of proteins that contain one or more characteristic EF-hand calcium-binding domains. This study describes and characterizes a novel EF-hand cDNA, CnidEF, from the sea anemone Anthopleura elegantissima (Phylum Cnidaria, Class Anthozoa). CnidEF was found to contain two EF-hand motifs near the C-terminus of the deduced amino acid sequence and two regions near the N-terminus that could represent degenerate EF-hand motifs. CnidEF homologues were also identified from two other sea anemone species. A combination of bioinformatic and molecular phylogenetic analyses was used to compare CnidEF to EF-hand proteins in other organisms. The closest homologues identified from these analyses were a luciferin binding protein (LBP) involved in the bioluminescence of the anthozoan Renilla reniformis, and a sarcoplasmic calcium-binding protein (SARC) involved in fluorescence of the annelid worm Nereis diversicolor. Predicted structure and folding analysis revealed a close association with bioluminescent aequorin (AEQ) proteins from the hydrozoan cnidarian Aequorea aequorea. Neighbor-joining analyses grouped CnidEF within the SARC lineage along with AEQ and other cnidarian bioluminescent proteins rather than in the lineage containing calmodulin (CAM) and troponin-C (TNC). -

Crystal Structure of Cardiac Troponin C Regulatory Domain in Complex with Cadmium and Deoxycholic Acid Reveals Novel Conformation
doi:10.1016/j.jmb.2011.08.049 J. Mol. Biol. (2011) 413, 699–711 Contents lists available at www.sciencedirect.com Journal of Molecular Biology journal homepage: http://ees.elsevier.com.jmb Crystal Structure of Cardiac Troponin C Regulatory Domain in Complex with Cadmium and Deoxycholic Acid Reveals Novel Conformation Alison Yueh Li 1, 2†, Jaeyong Lee 1†, Dominika Borek 3, Zbyszek Otwinowski 3, Glen F. Tibbits 1, 2, 4 and Mark Paetzel 1, 2⁎ 1Department of Molecular Biology and Biochemistry, Simon Fraser University, South Science Building, 8888 University Drive, Burnaby, British Columbia, Canada V5A 1S6 2Department of Biomedical Physiology and Kinesiology, Molecular Cardiac Physiology Group, Simon Fraser University, 8888 University Drive, Burnaby, British Columbia, Canada V5A 1S6 3Department of Biochemistry, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard, Dallas, TX 75390, USA 4Cardiovascular Sciences, Child and Family Research Institute, 950 West 28th Avenue, Vancouver, BC, Canada V5Z 4H4 Received 20 June 2011; The amino-terminal regulatory domain of cardiac troponin C (cNTnC) plays received in revised form an important role as the calcium sensor for the troponin complex. Calcium 23 August 2011; binding to cNTnC results in conformational changes that trigger a cascade of accepted 24 August 2011 events that lead to cardiac muscle contraction. The cardiac N-terminal Available online domain of TnC consists of two EF-hand calcium binding motifs, one of which 6 September 2011 is dysfunctional in binding calcium. Nevertheless, the defunct EF-hand still maintains a role in cNTnC function. For its structural analysis by X-ray Edited by R. -

Emerging Roles of the Single EF-Hand Ca2+ Sensor Tescalcin in the Regulation of Gene Expression, Cell Growth and Differentiation Ksenia G
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 521 doi:10.1242/jcs.200378 CORRECTION Correction: Emerging roles of the single EF-hand Ca2+ sensor tescalcin in the regulation of gene expression, cell growth and differentiation Ksenia G. Kolobynina, Valeria V. Solovyova, Konstantin Levay, Albert A. Rizvanov and Vladlen Z. Slepak There was an error published in J. Cell Sci. 129, 3533–3540. Incorrect language was used in the Commentary that concerns the apparent controversy between the results of tescalcin knockdown in hematopoietic precursor cell lines and its gene knockout in mice reported by Ukarapong et al. (2012). In the Commentary, the wording is as follows: “To explain the conflicting data, the authors [Ukarapong et al.] speculate that CHP1 and/or CHP2 could compensate for the lack of tescalcin, or that their genetic knockout was incomplete and allowed for the expression of a partial protein (Ukarapong et al., 2012)”. This gives the impression that Ukarapong et al. themselves argued about the incomplete knockout. The wording should read: “To explain the conflicting data, the authors [Ukarapong et al.] speculate that CHP1 and/or CHP2 could compensate for the lack of tescalcin (Ukarapong et al., 2012). Another possibility can be associated with the presence of a transcript corresponding to exons 1, 2, 7 and 8 of the TESC gene in the knockout mouse tissues detected by Ukarapong et al. Although unlikely, this finding leaves a formal possibility for this mRNA to be translated into a product with residual functional activity”. The authors of the Commentary apologize to Ukarapong et al. -

Key Protein Alterations Associated with Hyperdynamic Cardiac Function
Hellenic J Cardiol 48: 30-36, 2007 Original Research Key Protein Alterations Associated with Hyperdynamic Cardiac Function: Insights Based on Proteomic Analysis of the Protein Phosphatase 1 Inhibitor-1 Overexpressing Hearts 1 1 1,2 ANAND PATHAK , BRENT BALDWIN , EVANGELIA G. KRANIAS 1Department of Pharmacology & Cell Biophysics, University of Cincinnati, College of Medicine, Cincinnati, Ohio USA; 2Biomedical Research Foundation of the Academy of Athens, Greece Key words: Protein Transgenesis based on organ specific gene expression has provided the basis to elucidate the functional role phosphatase 1 of proteins for the past 15 years. Using this technology, we showed that inhibition of the protein phos- inhibitor-1, proteomics, transgenesis, phatase 1, by its constitutively active inhibitor-1, significantly increases cardiac contractility and calcium metabolism. handling. To uncover protein changes accompanying the chronic increases in cardiac function of these transgenic hearts, we analyzed the cardiac proteome. Interestingly, we found significant increases in the lev- els of 6 proteins involved in metabolism, calcium binding and scavenging of oxido-reductive stress. These proteins were identified as: hydroxyacyl CoA dehydrogenase II, alpha subunit of the mitochondrial proton ATPase, peroxiredoxin 2, a novel EF-hand containing protein-2, annexin 5, and a previously uncharacterized cDNA. Thus, long-term cardiac specific overexpression of the protein phosphatase 1 inhibitor-1 and the as- sociated increases in cardiac contractility appear to herald changes in a rather small number of proteins, which may reflect important compensatory adaptations in a hyperdynamic heart. Manuscript received: December 4, 2006; Accepted: February 5, 2007. eversible protein phosphorylation coplasmic reticulum calcium cycling and and dephosphorylation mediated cardiomyocyte relaxation. -

Chapter 4 Calsequestrin
Chapter 4 Calsequestrin DAVID H. MacLENNAN Banting and Best Department of Medical Research C. H. Best Institute, University of Toronto Toronto, Ontario, Canada KEVIN P. CAMPBELL Department of Physiology and Biophysics University of Iowa Iowa City, Iowa and REINHART A. F. REITHMEIER Department of Biochemistry University of Alberta Edmonton, Alberta, Canada I. Sarcoplasmic Reticulum ............................................................................. 152 II. Isolated Sarcoplasmic Reticulum Vesicles ................................................. 153 III. Isolation of Calsequestrin ........................................................................... 155 IV. Ca2+ Binding by Calsequestrin ................................................................... 155 V. Size and Shape of Calsequestrin................................................................. 157 VI. Amino Acid Sequence................................................................................ 159 VII. Carbohydrate Content................................................................................. 160 VIII. Stains-All Staining...................................................................................... 160 IX. Properties of Calsequestrin in Other Muscles............................................. 161 X. Phosphorylation of Calsequestrin ............................................................... 162 XI. Localization of Calsequestrin ..................................................................... 163 XII. Biosynthesis............................................................................................... -

A Role for EF-Hand Proteins?
Review TRENDS in Microbiology Vol.10 No.2 February 2002 87 52 Wasylnka, J.A. and Moore, M.M. (2000) Adhesion factor of ras Cdc25p and controls the cAMP immunodominant β-galacto-furanose residue of Aspergillus species to extracellular matrix pathway in Saccharomyces cerevisiae. Mol. is carried by a novel glycosylinositol proteins: evidence for involvement of negative Microbiol. 30, 855–864 phosphorylceramide antigen. Biochemistry charged carbohydrates on the conidial surface. 60 Pereira, M. et al. (2000) Molecular cloning and 37, 8764–8775 Infect. Immun. 68, 3377–3384 characterization of a glucan synthase gene from 67 Toledo, M.S. et al. (1999) Characterization of 53 San Blas, G. and San Blas, F. (1993) Biochemistry the human pathogenic fungus Paracoccidioides sphingolipids from mycopathogens: factors of P. brasiliensis dimorphism. In brasiliensis. Yeast 165, 451–462 correlating with expression of 2-hydroxy fatty acyl Paracoccidioidomycosis (Franco, M. et al., eds), 61 Hogan, L.H. and Klein, B.S. (1994) Altered (E)-∆ [3]-unsaturation in cerebrosides of pp. 49–66, CRC Press expression of surface α-1,3-glucan in genetically Paracoccidioides brasiliensis and Aspergillus 54 Brown, A.J.P. and Gow, N.A.R. (1999) Regulatory related strains of Blastomyces dermatitidis that fumigatus. Biochemistry 38, 7294–7306 networks controlling Candida albicans differ in virulence. Infect. Immun. 62, 3543–3546 68 Toledo, M.S. et al. (1995) Glycolipids from morphogenesis. Trends Microbiol. 7, 333–338 62 Katayama, S. et al. (1999) Fission yeast α-glucan Paracoccidioides brasiliensis. Isolation of a 55 Borges-Walmsley, M.I. and Walmsley, A.R. (2000) synthase Mok1 requires the actin cytoskeleton to galactofuranose-containing glycolipid reactive cAMP signalling in pathogenic fungi: control of localize the sites of growth and plays an essential with sera of patients with paracoccidioidomycosis.