Investigating the Mechanism of Action of Hormones Used in Hormone Replacement Therapy Via Estrogen Receptor Subtypes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
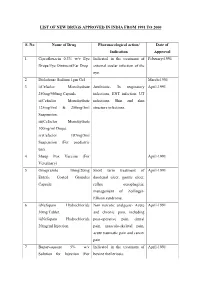
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

Comparing the Effects of Combined Oral Contraceptives Containing Progestins with Low Androgenic and Antiandrogenic Activities on the Hypothalamic-Pituitary-Gonadal Axis In
JMIR RESEARCH PROTOCOLS Amiri et al Review Comparing the Effects of Combined Oral Contraceptives Containing Progestins With Low Androgenic and Antiandrogenic Activities on the Hypothalamic-Pituitary-Gonadal Axis in Patients With Polycystic Ovary Syndrome: Systematic Review and Meta-Analysis Mina Amiri1,2, PhD, Postdoc; Fahimeh Ramezani Tehrani2, MD; Fatemeh Nahidi3, PhD; Ali Kabir4, MD, MPH, PhD; Fereidoun Azizi5, MD 1Students Research Committee, School of Nursing and Midwifery, Department of Midwifery and Reproductive Health, Shahid Beheshti University of Medical Sciences, Tehran, Islamic Republic Of Iran 2Reproductive Endocrinology Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Islamic Republic Of Iran 3School of Nursing and Midwifery, Department of Midwifery and Reproductive Health, Shahid Beheshti University of Medical Sciences, Tehran, Islamic Republic Of Iran 4Minimally Invasive Surgery Research Center, Iran University of Medical Sciences, Tehran, Islamic Republic Of Iran 5Endocrine Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Islamic Republic Of Iran Corresponding Author: Fahimeh Ramezani Tehrani, MD Reproductive Endocrinology Research Center Research Institute for Endocrine Sciences Shahid Beheshti University of Medical Sciences 24 Parvaneh Yaman Street, Velenjak, PO Box 19395-4763 Tehran, 1985717413 Islamic Republic Of Iran Phone: 98 21 22432500 Email: [email protected] Abstract Background: Different products of combined oral contraceptives (COCs) can improve clinical and biochemical findings in patients with polycystic ovary syndrome (PCOS) through suppression of the hypothalamic-pituitary-gonadal (HPG) axis. Objective: This systematic review and meta-analysis aimed to compare the effects of COCs containing progestins with low androgenic and antiandrogenic activities on the HPG axis in patients with PCOS. -

Properties and Units in Clinical Pharmacology and Toxicology
Pure Appl. Chem., Vol. 72, No. 3, pp. 479–552, 2000. © 2000 IUPAC INTERNATIONAL FEDERATION OF CLINICAL CHEMISTRY AND LABORATORY MEDICINE SCIENTIFIC DIVISION COMMITTEE ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)# and INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY CHEMISTRY AND HUMAN HEALTH DIVISION CLINICAL CHEMISTRY SECTION COMMISSION ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)§ PROPERTIES AND UNITS IN THE CLINICAL LABORATORY SCIENCES PART XII. PROPERTIES AND UNITS IN CLINICAL PHARMACOLOGY AND TOXICOLOGY (Technical Report) (IFCC–IUPAC 1999) Prepared for publication by HENRIK OLESEN1, DAVID COWAN2, RAFAEL DE LA TORRE3 , IVAN BRUUNSHUUS1, MORTEN ROHDE1, and DESMOND KENNY4 1Office of Laboratory Informatics, Copenhagen University Hospital (Rigshospitalet), Copenhagen, Denmark; 2Drug Control Centre, London University, King’s College, London, UK; 3IMIM, Dr. Aiguader 80, Barcelona, Spain; 4Dept. of Clinical Biochemistry, Our Lady’s Hospital for Sick Children, Crumlin, Dublin 12, Ireland #§The combined Memberships of the Committee and the Commission (C-NPU) during the preparation of this report (1994–1996) were as follows: Chairman: H. Olesen (Denmark, 1989–1995); D. Kenny (Ireland, 1996); Members: X. Fuentes-Arderiu (Spain, 1991–1997); J. G. Hill (Canada, 1987–1997); D. Kenny (Ireland, 1994–1997); H. Olesen (Denmark, 1985–1995); P. L. Storring (UK, 1989–1995); P. Soares de Araujo (Brazil, 1994–1997); R. Dybkær (Denmark, 1996–1997); C. McDonald (USA, 1996–1997). Please forward comments to: H. Olesen, Office of Laboratory Informatics 76-6-1, Copenhagen University Hospital (Rigshospitalet), 9 Blegdamsvej, DK-2100 Copenhagen, Denmark. E-mail: [email protected] Republication or reproduction of this report or its storage and/or dissemination by electronic means is permitted without the need for formal IUPAC permission on condition that an acknowledgment, with full reference to the source, along with use of the copyright symbol ©, the name IUPAC, and the year of publication, are prominently visible. -

2010 Prohibited List
The World Anti-Doping Code THE 2010 PROHIBITED LIST INTERNATIONAL STANDARD The official text of the Prohibited List shall be maintained by WADA and shall be published in English and French. In the event of any conflict between the English and French versions, the English version shall prevail. This List shall come into effect on 1 January 2010 The Prohibited List 2010 19 September 2009 THE 2010 PROHIBITED LIST WORLD ANTI-DOPING CODE Valid 1 January 2010 All Prohibited Substances shall be considered as “Specified Substances” except Substances in classes S1, S2.1 to S2.5, S.4.4 and S6.a, and Prohibited Methods M1, M2 and M3. SUBSTANCES AND METHODS PROHIBITED AT ALL TIMES (IN- AND OUT-OF-COMPETITION) PROHIBITED SUBSTANCES S1. ANABOLIC AGENTS Anabolic agents are prohibited. 1. Anabolic Androgenic Steroids (AAS) a. Exogenous* AAS, including: 1-androstendiol (5α-androst-1-ene-3β,17β-diol ); 1-androstendione (5α- androst-1-ene-3,17-dione); bolandiol (19-norandrostenediol); bolasterone; boldenone; boldione (androsta-1,4-diene-3,17-dione); calusterone; clostebol; danazol (17α-ethynyl-17β-hydroxyandrost-4-eno[2,3-d]isoxazole); dehydrochlormethyltestosterone (4-chloro-17β-hydroxy-17α-methylandrosta- 1,4-dien-3-one); desoxymethyltestosterone (17α-methyl-5α-androst-2-en- 17β-ol); drostanolone; ethylestrenol (19-nor-17α-pregn-4-en-17-ol); fluoxymesterone; formebolone; furazabol (17β-hydroxy-17α-methyl-5α- androstano[2,3-c]-furazan); gestrinone; 4-hydroxytestosterone (4,17β- dihydroxyandrost-4-en-3-one); mestanolone; mesterolone; metenolone; methandienone -

Giving Another Chance to Mifepristone in Pharmacotherapy for Aggressive Meningiomas—A Likely Synergism with Hydroxyurea?
Curr Probl Cancer 40 (2016) 229–243 Contents lists available at ScienceDirect Curr Probl Cancer journal homepage: www.elsevier.com/locate/cpcancer Giving another chance to mifepristone in pharmacotherapy for aggressive meningiomas—A likely synergism with hydroxyurea? İlhan Elmaci, MDa, Meric A. Altinoz, MDb,*, Aydin Sav, MDc, Zeliha Yazici, PhDd, Aysel Ozpinar, PhDe Introduction Meningiomas: An underestimated health problem Meningiomas are the most frequently reported intracranial tumors, accounting for approx- imately one-fourth of all reported primary brain neoplasms.1 They are benign in approximately 90% of the cases and the remaining cases are either borderline or atypical (World Health Organization [WHO] grade II) or malignant (WHO grade III).1 In the United States, the incidence rates with similar age standardization estimated from figures provided by the Central Brain Tumor Registrywere1.8formenand4.2per100,000forwomenin2006.1 Increasing incidence rates of meningiomas have been reported from several industrialized countries since the early 1980s. But recent studies revealed that real meningioma incidence is even higher.1 As meningiomas are mostly benign, they are not covered by most cancer registries.1 Nevertheless, in Finland, as in other Nordic countries, all surgeons and pathologists are obliged to report all tumors of the central nervous system, both malignant and benign, to the cancer registry.1 To reveal the real incidence of Grant/Financial Support: none. This is original work and is not under consideration for publication elsewhere. This submission is for the special issue being compiled by Beata Holkova on the topic Amylodosis. a Department of Neurosurgery, Memorial Hospital, Istanbul, Turkey b Department of Immunology, Experimental Medical Research Institute/DETAE, Istanbul University, Istanbul, Turkey c Nisantasi Neuropathology Group, Istanbul, Turkey d Department of Pharmacology, Cerrahpasa Faculty of Medicine, Istanbul University, Turkey e Department of Biochemistry, Acibadem University, Istanbul, Turkey * Corresponding author: Meric A. -

Progestogens and Venous Thromboembolism Among Postmenopausal Women Using Hormone Therapy. Marianne Canonico, Geneviève Plu-Bureau, Pierre-Yves Scarabin
Progestogens and venous thromboembolism among postmenopausal women using hormone therapy. Marianne Canonico, Geneviève Plu-Bureau, Pierre-Yves Scarabin To cite this version: Marianne Canonico, Geneviève Plu-Bureau, Pierre-Yves Scarabin. Progestogens and venous throm- boembolism among postmenopausal women using hormone therapy.. Maturitas, Elsevier, 2011, 70 (4), pp.354-60. 10.1016/j.maturitas.2011.10.002. inserm-01148705 HAL Id: inserm-01148705 https://www.hal.inserm.fr/inserm-01148705 Submitted on 5 May 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Progestogens and VTE Finale version Progestogens and venous thromboembolism among postmenopausal women using hormone therapy Marianne Canonico1,2, Geneviève Plu-Bureau1,3 and Pierre-Yves Scarabin1,2 1 Centre for Research in Epidemiology and Population Health, U1018, Hormones and Cardiovascular Disease 2 University Paris-Sud, UMR-S 1018, Villejuif, France 3 University Paris Descartes and Hôtel-Dieu Hospital, Paris, France Adresse: 16 av. Paul Vaillant Couturier 94807 Villejuif Cedex Tel: +33 1 45 59 51 66 Fax: +33 1 45 59 51 70 Corresponding author: Marianne Canonico ([email protected]) 1/21 Progestogens and VTE Finale version Abstract Hormone therapy (HT) is the most effective treatment for correcting menopausal symptoms after menopause. -

In Vivo Effect of PC-SPES on Prostate Growth and Hepatic CYP3A Expression in Rats
JPET Fast Forward. Published on April 3, 2003 as DOI: 10.1124/jpet.102.048645 JPETThis Fast article Forward. has not been Published copyedited and on formatted. April 3, The 2003 final asversion DOI:10.1124/jpet.102.048645 may differ from this version. JPET #48645 In Vivo Effect of PC-SPES on Prostate Growth and Hepatic CYP3A Expression in Rats Teri Wadsworth, Hataya Poonyagariyagorn, Elinore Sullivan, Dennis Koop and Charles E. Roselli. Downloaded from Department of Physiology and Pharmacology Oregon Health & Science University, Portland, OR. jpet.aspetjournals.org at ASPET Journals on September 26, 2021 1 Copyright 2003 by the American Society for Pharmacology and Experimental Therapeutics. JPET Fast Forward. Published on April 3, 2003 as DOI: 10.1124/jpet.102.048645 This article has not been copyedited and formatted. The final version may differ from this version. JPET #48645 Running Title: In vivo effects of PC-SPES Correspondence: Dr. Charles E. Roselli, Department of Physiology and Pharmacology L334, Oregon Health Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR 97201-3098, Tel (503) 494-5837, FAX (503) 494-4352, email: [email protected] Number of text pages: 20 number of tables: 4 Downloaded from number of figures: 4 number of references: 40 jpet.aspetjournals.org number of words in the Abstract: 250 number of words the Introduction: 748 number of words in the Discussion: 1478 at ASPET Journals on September 26, 2021 nonstandard abbreviations: National Institute of Diabetes and Digestive and Kidney Disease (NIDDK); -

Contraceptive Choices for Women with Inflammatory Bowel Disease
J Fam Plann Reprod Health Care: first published as 10.1783/147118903101197782 on 1 July 2003. Downloaded from Faculty of Family Planning and Reproductive Health Care Clinical Effectiveness Unit A unit funded by the FFPRHC and supported by the University of Aberdeen and SPCERH to provide guidance on evidence-based practice FFPRHC Guidance (July 2003) Contraceptive Choices for Women with Inflammatory Bowel Disease Journal of Family Planning and Reproductive Health Care 2003; 29(3): 127–135 This Guidance provides information on the effects of inflammatory bowel disease (IBD) in women of reproductive age with particular reference to contraceptive choices, fertility and pregnancy. For comprehensive advice on the management of patients with IBD, readers should refer to guidelines from the British Society for Gastroenterology.1 A key to the grades of recommendations, based on levels of evidence, is given at the end of this document. Details of the methods used by the Clinical Effectiveness Unit (CEU) in developing this Guidance, and evidence tables summarising the research basis of the recommendations, are available on the Faculty website (www.ffprhc.org.uk). Abbreviations used include: 5-aminosalicylic acid (5-ASA), body mass index (BMI), bone mineral density (BMD), combined oral contraception (COC), confidence interval (CI), copper-bearing intrauterine contraceptive device (IUD), Crohn’s disease (CD), depot medroxyprogesterone acetate (DMPA), dual X-ray absorptiometry (DEXA), emergency contraception (EC), gastrointestinal (GI), incidence rate ratio (IRR), inflammatory bowel disease (IBD), levonorgestrel-releasing intrauterine system (IUS), odds ratio (OR), progestogen-only emergency contraception (POEC), progestogen-only pill (POP), anti-tumour necrosis factor-alpha (anti- TNF-a), ulcerative colitis (UC), venous thromboembolism (VTE), World Health Organization (WHO). -

CHEQUE® DROPS CII Pharmacia & Upjohn Estrous Preventive Mibolerone Drops NADA No.: 102-709 Active Ingredient(S): Each Ml Co
CHEQUE® DROPS CII Pharmacia & Upjohn Estrous Preventive Mibolerone drops NADA No.: 102-709 Active Ingredient(s): Each mL contains 100 mcg mibolerone and propylene glycol, qs. Indications: CHEQUE® (mibolerone) Drops are administered orally for estrous (heat) prevention in adult female dogs not intended primarily for breeding purposes. CHEQUE® Drops dosage should be discontinued after 24 months of use. CHEQUE® Drops are effective in preventing estrus only when drug administration is initiated 30 days prior to the start of the proestrus. It should not be used in an attempt to abbreviate an estrous period. It should not be used in bitches prior to the first estrous period. Pharmacology: CHEQUE® (mibolerone) Drops are 17-b-hydroxy-7a, 17-dimethyl-estr-4-en-3-one, generic name mibolerone, a non-progestational steroid. In its pure form, mibolerone is a white crystalline solid. The compound is stable under ordinary conditions and temperatures. Actions: More than 90 percent of normal, mature cycling bitches did not exhibit estrus when administered CHEQUE® (mibolerone) Drops in adequate doses. Mibolerone has been shown to be an anabolic and androgenic steroid in rats. When compared to methyltestosterone, it is 41 times more potent as an anabolic agent and 16 times more potent as an androgen. Mibolerone has no estrogenic activity in mice when used at levels up to 1.0 mg per animal per day. It is antiestrogenic in mice in intravaginal tests at 0.9 mcg and in subcutaneous tests at 10 mcg or less when tested against estradiol. In the bitch, mibolerone has androgenic, anabolic and antigonadotrophic activity. -

Malta Medicines List April 08
Defined Daily Doses Pharmacological Dispensing Active Ingredients Trade Name Dosage strength Dosage form ATC Code Comments (WHO) Classification Class Glucobay 50 50mg Alpha Glucosidase Inhibitor - Blood Acarbose Tablet 300mg A10BF01 PoM Glucose Lowering Glucobay 100 100mg Medicine Rantudil® Forte 60mg Capsule hard Anti-inflammatory and Acemetacine 0.12g anti rheumatic, non M01AB11 PoM steroidal Rantudil® Retard 90mg Slow release capsule Carbonic Anhydrase Inhibitor - Acetazolamide Diamox 250mg Tablet 750mg S01EC01 PoM Antiglaucoma Preparation Parasympatho- Powder and solvent for solution for mimetic - Acetylcholine Chloride Miovisin® 10mg/ml Refer to PIL S01EB09 PoM eye irrigation Antiglaucoma Preparation Acetylcysteine 200mg/ml Concentrate for solution for Acetylcysteine 200mg/ml Refer to PIL Antidote PoM Injection injection V03AB23 Zovirax™ Suspension 200mg/5ml Oral suspension Aciclovir Medovir 200 200mg Tablet Virucid 200 Zovirax® 200mg Dispersible film-coated tablets 4g Antiviral J05AB01 PoM Zovirax® 800mg Aciclovir Medovir 800 800mg Tablet Aciclovir Virucid 800 Virucid 400 400mg Tablet Aciclovir Merck 250mg Powder for solution for inj Immunovir® Zovirax® Cream PoM PoM Numark Cold Sore Cream 5% w/w (5g/100g)Cream Refer to PIL Antiviral D06BB03 Vitasorb Cold Sore OTC Cream Medovir PoM Neotigason® 10mg Acitretin Capsule 35mg Retinoid - Antipsoriatic D05BB02 PoM Neotigason® 25mg Acrivastine Benadryl® Allergy Relief 8mg Capsule 24mg Antihistamine R06AX18 OTC Carbomix 81.3%w/w Granules for oral suspension Antidiarrhoeal and Activated Charcoal -

SNI May-Jun-2011 Cover Final
OPEN ACCESS Editor-in-Chief: Surgical Neurology International James I. Ausman, MD, PhD For entire Editorial Board visit : University of California, Los http://www.surgicalneurologyint.com Angeles, CA, USA Review Article Stuck at the bench: Potential natural neuroprotective compounds for concussion Anthony L. Petraglia, Ethan A. Winkler1, Julian E. Bailes2 Department of Neurosurgery, University of Rochester Medical Center, Rochester, NY, 1University of Rochester School of Medicine and Dentistry, Rochester, NY, 2Department of Neurosurgery, North Shore University Health System, Evanston, IL, USA E-mail: *Anthony L. Petraglia - [email protected]; Ethan A. Winkler - [email protected]; Julian E. Bailes - [email protected] *Corresponding author Received: 29 August 11 Accepted: 22 September 11 Published: 12 October 11 This article may be cited as: Petraglia AL, Winkler EA, Bailes JE. Stuck at the bench: Potential natural neuroprotective compounds for concussion. Surg Neurol Int 2011;2:146. Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2011/2/1/146/85987 Copyright: © 2011 Petraglia AL. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Background: While numerous laboratory studies have searched for neuroprotective treatment approaches to traumatic brain injury, no therapies have successfully translated from the bench to the bedside. Concussion is a unique form of brain injury, in that the current mainstay of treatment focuses on both physical and cognitive rest. Treatments for concussion are lacking. The concept of neuro-prophylactic compounds or supplements is also an intriguing one, especially as we are learning more about the relationship of numerous sub-concussive blows and/or repetitive concussive impacts and the development of chronic neurodegenerative disease. -

Rominder P.S. Suri, Phd, PE Robert F. Chimchirian & Sandhya Meduri Department of Civil & Environmental Engineering Villa
Emerging Contaminants and Water Supply – Speaker Abstracts Emerging Contaminants and Water Supply – Speaker Abstracts PRESENCE OF ESTROGEN HORMONES AND ANTIBIOTICS IN detection limits for all 12 estrogen compounds ranged from 0.1 ng L-1 to THE ENVIRONMENT 10 ng L-1 using a sample size of 1 L. Eight antibiotics and venlaflaxine Rominder P.S. Suri, PhD, PE were detected in the effluent with concentrations that ranged from 50 to Robert F. Chimchirian & Sandhya Meduri 2,930 ng L-1. Department of Civil & Environmental Engineering Twenty-one streams in Chester County, southeast Pennsylvania, were Villanova University, Villanova PA 19085 also sampled for aqueous concentrations of the above hormones and 610-519-7631 [email protected] antibiotics. Chester County represents a very diverse land use. In addition to concentrated urban developments, there are many In recent years, the detection of trace amount of pharmaceutically active agricultural farms (dairy, cattle, hog, poultry, and mushroom). The compounds (PACs) in the surface water has gained widespread watershed is comprised of about 280,000 acres that eventually drain into attention. The presence of the PACs in the natural water systems causes the Chesapeake Bay via the Susquehanna River. The results showed that ecological problems and possible adverse effects to humans. Two classes there was at least one estrogen detected in every stream sampled and six of PACs, estrogen hormones and antibiotics make their way into the estrogens were detected at three different locations. The most abundant surface waters through effluents from wastewater treatment plants and PAC was estrone, which was detected in all but one stream, with areas where animal manure and biosolids are land applied, amongst concentrations as high as 2.4 ng L-1.