Immune Evasion MHC Class II Processing Pathway for Herpes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 ICAM3-Fc Outperforms Receptor-Specific Antibodies
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 10 April 2019 doi:10.20944/preprints201904.0118.v1 Peer-reviewed version available at Molecules 2019, 24, 1825; doi:10.3390/molecules24091825 ICAM3-Fc outperforms receptor-specific antibodies targeted nanoparticles to dendritic cells for cross-presentation Luis J. Cruz,1* Paul J. Tacken,2 Johan S. van der Schoot,2 Felix Rueda,3 Ruurd Torensma,2 Carl G. Figdor2* 1Translational Nanobiomaterials and Imaging, Department of Radiology, Leiden University Medical Center, Albinusdreef 2, 2333 ZA Leiden, The Netherlands. 2Department of Tumor Immunology, Radboud Insititute for Molecular Life Sciences, Radboud University Medical Center, Postbox 9101, 6500 HB Nijmegen, The Netherlands. 3Department of Biochemistry and Molecular Biology, University of Barcelona, Diagonal 643, 08028 Barcelona, Spain. Running title: ICAM3-Fc- versus antibody-targeted NP vaccines to human DCs Keywords: Delivery system; nanoparticle; targeting. AUTHOR INFORMATION Corresponding Author *Dr. Luis J. Cruz Translational Nanobiomaterials and Imaging, Department of Radiology, Bldg.1, C5-60. Leiden University Medical Center, Albinusdreef 2 2333 ZA Leiden, The Netherlands Tel: +31 71 5263025 Email: [email protected] *Prof. Dr. Carl G. Figdor Department of Tumor Immunology, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre, Postbox 9101, 6500 HB Nijmegen, The Netherlands. Fax: +31-24-3540339; Tel.: +31-24-3617600; E-mail: [email protected] 1 © 2019 by the author(s). Distributed -

Class II MHC Cytoplasmic Domain-Mediated Signaling in B Cells a Tail of Two Signals
Human Immunology 80 (2019) 32–36 Contents lists available at ScienceDirect Human Immunology journal homepage: www.elsevier.com/locate/humimm Class II MHC cytoplasmic domain-mediated signaling in B cells: A tail of two T signals Jonathan A. Harton Department of Immunology & Microbial Disease, Albany Medical College, 47 New Scotland Avenue, MC-151, Albany, NY 12208, USA ARTICLE INFO ABSTRACT Keywords: In addition to their role in antigen presentation, class II MHC molecules also transmit signals to B lymphocytes. MHC class II Class II MHC-mediated signals initiate a range of events in B cells, including induction of cell surface proteins, HLA-D initiation of cell-cycle progression/proliferation, activation of or protection from apoptosis, and antigen-de- Signaling pendent plasma cell differentiation. Although various transmembrane signaling proteins associate with classII B cells MHC molecules, the class II MHC cytoplasmic domains are essential for signals leading to increased intracellular cAMP and activation of protein kinase C (PKC). Although truncation and mutagenesis studies have provided considerable information about the cytoplasmic domain sequences required, how class II MHC molecules elicit cAMP and PKC activation is not known. Further, appropriate T-dependent B cell responses require intact cAMP and PKC signaling, but the extent to which class II MHC signals are involved is also unknown. This review details our current knowledge of class II MHC cytoplasmic domain signaling in B cells with an emphasis on the likely importance of class II MHC signals for T-dependent antibody responses. 1. Introduction complex, is only found on CD4+ T cells which are dependent upon thymic MHC class II for their development. -

Cell Biology from an Immune Perspective in This Lecture We Will
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Cell Biology from an Immune Perspective In this lecture we will very briefly review some aspects of cell biology which are required as background knowledge in order to understand how the immune system works. These will include: 1. A brief overview of protein trafficking 2. Signal transduction 3. The cell cycle Some of these issues will be treated in greater depth in later lectures. Protein Trafficking/The Secretory Pathway: From an immune perspective the secretory compartment and structures enclosed by vesicles are “seen” in different ways from proteins that reside in the cytosol or the nucleus. We will briefly review the secretory and endocytic pathways and discuss the biogenesis of membrane proteins. Some of the issues that will be discussed are summarized in Figures 1-3. Early endosomes Late endosomes Lysosomes Golgi Vesiculo-Tubular Compartment ER Figure 1. An overview of the secretory pathway Early endosomes Late endosomes Multivesicular and multilamellar bodies Golgi ER Proteasomes Figure 2. Protein degradation occurs mainly in lysosomes and proteasomes Proteins that enter the cell from the environment are primarily degraded in lysosomes. Most cytosolic and nuclear proteins are degraded in organelles called proteasomes. Intriguingly these two sites of degradation are each functionally linked to distinct antigen presentation pathways, different kinds of MHC molecules and the activation of different categories of T cells. Integral membrane proteins maybe inserted into the membrane in a number of ways, the two most common of these ways being considered in Figure 3. -

MHC Class II Stabilization at the Surface of Human Dendritic Cells Is the Result of Maturation-Dependent MARCH I Down-Regulation
MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation Aude De Gassart*†‡, Voahirana Camosseto*†‡, Jacques Thibodeau§¶, Maurizio Ceppi*†‡, Nadia Catalan*†‡, Philippe Pierre*†‡ʈ, and Evelina Gatti*†‡ʈ *Centre d’Immunologie de Marseille–Luminy, Universite´ delaMe´ diterrane´e, Case 906, 13288 Marseille Cedex 9, France; †Institut National de la Sante´etde la Recherche Me´dicale, Unite´631, 13288 Marseille, France; ‡Centre National de la Recherche Scientifique, Unite´Mixte de Recherche 6102, 13288 Marseille, France; §De´partement de Microbiologie et Immunologie, Universite´de Montre´al, Montreal, QC, Canada H3T 1J4; and ¶Institut National de la Sante´et de la Recherche Me´dicale, Unite´743, Montreal, QC, Canada H3T 1J4 Edited by Ralph M. Steinman, The Rockefeller University, New York, NY, and approved January 9, 2008 (received for review September 19, 2007) In response to Toll-like receptor ligands, dendritic cells (DCs) dramatically when cells are exposed to LPS (12, 13). As determined dramatically enhance their antigen presentation capacity by sta- by confocal microscopy, iDCs accumulate HLA-DR molecules in bilizing at the cell-surface MHC II molecules. We demonstrate here LAMP1ϩ late endosomes and lysosomes. On activation by LPS, that, in human monocyte-derived DCs, the RING-CH ubiquitin E3 rapid relocalization of MHC II to the cell surface is observed (4–8 ligase, membrane-associated RING-CH I (MARCH I), promotes the h) [supporting information (SI) Fig. 7A]. Recently, this process has ubiquitination of the HLA-DR -chain. Thus, in nonactivated DCs, been shown to be associated with a loss of ubiquitination on the MARCH I induces the surface internalization of mature HLA-DR class II -chain in mouse bone marrow-derived DCs (bmDCs) (4, complexes, therefore reducing their stability and levels. -

Interaction Between CD8 and Major Histocompatibility Complex (MHC) Class I Mediated by Multiple Contact Surfaces That Include Th
Interaction Between CD8 and Major Histocompatibility Complex (MHC) Class I Mediated by Multiple Contact Surfaces that Include the oL2 and oL3 Domains of MHC Class I By Jiaren Sun,* DanielJ. Leahy,~ and Paula B. Kavathas*w From the *Department of Laboratory Medicine and Section of lmmunobiology, Yale University School of Medicine, New Haven, Connecticut 06520-8035; "Ji.Departmentof Biophysics and Biophysical Chemistry,Johns Hopkins University School of Medicine, Baltimore, Maryland 21205-2185; wDepartment of Genetics, Yale University School of Medicine, New Haven, Connecticut 06520-8035 Summary Downloaded from http://rupress.org/jem/article-pdf/182/5/1275/1107333/1275.pdf by guest on 03 October 2021 The cell surface glycoprotein CD8 functions as a coreceptor with the TCR on cytotoxic T lymphocytes. Mutational analysis of the binding site of CD8 for MHC class I predicted that distinct surfaces of CD8 would interact with both the a2 and a3 domains of class I. Using a cell-cell adhesion assay, we identified three residues Q115, D122, and E128 in the or2 domain of class I critical for interaction with CD8. The side chains of these residues point towards a cavity formed by the 0tl/e~2 platform, the or3 domain and [32-microglobulin ([32m) of class I. These residues were predicted to contact CD8 based on a bivalent model of interaction be- tween one CD8o~/oL homodimer and two MHC class I molecules. These results therefore pro- vide support for the model. HC class I molecules are highly polymorphic pro- critical residues in the 0~2 domain, two located underneath M teins that bind antigenic peptides and present them the peptide-binding floor and one on a nearby loop, all to T cells. -

Footprints of Antigen Processing Boost MHC Class II Natural Ligand
Barra et al. Genome Medicine (2018) 10:84 https://doi.org/10.1186/s13073-018-0594-6 RESEARCH Open Access Footprints of antigen processing boost MHC class II natural ligand predictions Carolina Barra1† , Bruno Alvarez1†, Sinu Paul2, Alessandro Sette2, Bjoern Peters2, Massimo Andreatta1, Søren Buus3 and Morten Nielsen1,4* Abstract Background: Major histocompatibility complex class II (MHC-II) molecules present peptide fragments to T cells for immune recognition. Current predictors for peptide to MHC-II binding are trained on binding affinity data, generated in vitro and therefore lacking information about antigen processing. Methods: We generate prediction models of peptide to MHC-II binding trained with naturally eluted ligands derived from mass spectrometry in addition to peptide binding affinity data sets. Results: We show that integrated prediction models incorporate identifiable rules of antigen processing. In fact, we observed detectable signals of protease cleavage at defined positions of the ligands. We also hypothesize a role of the length of the terminal ligand protrusions for trimming the peptide to the MHC presented ligand. Conclusions: The results of integrating binding affinity and eluted ligand data in a combined model demonstrate improved performance for the prediction of MHC-II ligands and T cell epitopes and foreshadow a new generation of improved peptide to MHC-II prediction tools accounting for the plurality of factors that determine natural presentation of antigens. Keywords: MHC-II, Binding predictions, Eluted ligands, T cell epitope, Neural networks, Antigen processing, Machine learning, Mass spectrometry Background MHC-II binding groove, unlike MHC class I, is open at Major histocompatibility complex class II (MHC-II) mole- both ends. -
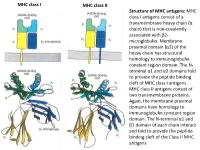
MHC Class I Antigens Consist of a Transmembrane Heavy Chain (Α Chain) That Is Non-Covalently Associated with Β2- Microglobulin
MHC class I MHC class II Structure of MHC antigens: MHC class I antigens consist of a transmembrane heavy chain (α chain) that is non-covalently associated with β2- microglobulin. Membrane proximal domain (α3) of the heavy chain has structural homology to immunoglobulin constant region domain. The N- terminal α1 and α2 domains fold to provide the peptide binding cleft of MHC class I antigens. MHC class II antigens consist of two transmembrane proteins. Again, the membrane proximal domains have homology to immunoglobulin constant region domain. The N-terminal α1 and β1 domain of each chain interact and fold to provide the peptide binding cleft of the Class II MHC antigens MHC class I MHC class II A top-down view of the peptide binding cleft of MHC antigens. The peptide binding cleft for MHC class I antigens is relatively narrower at the ends and accommodate 8- 10 amino acid residues long peptides. In contrast, due to non-covalent association between the N-terminal domains of the two chains of the class II antigens, the peptide binding cleft is open at the ends. This permits binding of larger peptides, typically 13-20 aa long. T cell receptor Interaction between T cell receptor and MHC-peptide complex: The T cell receptors binds to the peptide-MHC complex in a diagonal fashion. The CDR1 and CDR2 regions of the α and β chains of the T cell receptor bind to the α-helical walls of the peptide binding cleft. The CDR3 regions on the other hand bind to the peptide present in the cleft. -

Fcγ Receptors As Regulators of Immune Responses
REVIEWS Fcγ receptors as regulators of immune responses Falk Nimmerjahn* and Jeffrey V. Ravetch‡ Abstract | In addition to their role in binding antigen, antibodies can regulate immune responses through interacting with Fc receptors (FcRs). In recent years, significant progress has been made in understanding the mechanisms that regulate the activity of IgG antibodies in vivo. In this Review, we discuss recent studies addressing the multifaceted roles of FcRs for IgG (FcγRs) in the immune system and how this knowledge could be translated into novel therapeutic strategies to treat human autoimmune, infectious or malignant diseases. Regulatory T cells A productive immune response results from the effec- of antigenic peptides that are presented on the surface A T cell subset that is capable tive integration of positive and negative signals that of DCs to cytotoxic T cells, T helper cells, and regulatory of suppressing the activity have an impact on both innate and adaptive immune T cells. Thus, FcγRs are involved in regulating a multitude of other antigen-specific T cells cells. When positive signals dominate, cell activation of innate and adaptive immune responses, which makes including autoreactive T cells. Depletion of regulatory and pro-inflammatory responses ensue, resulting in the them attractive targets for the development of novel T cells results in the loss of elimination of pathogenic microorganisms and viruses. immunotherapeutic approaches (FIG. 1). peripheral tolerance and the In the absence of such productive stimulation, cell activa- In this Review, we summarize our current under- development of autoimmune tion is blocked and active anti-inflammatory responses standing of the role of different members of the FcγR disease. -

Class II Major Histocompatibility Complex Mutant Mice to Study The
Class II major histocompatibility complex mutant mice PNAS PLUS to study the germ-line bias of T-cell antigen receptors Daniel Silbermana,b, Sai Harsha Krovib, Kathryn D. Tuttleb, James Crooksc, Richard Reisdorphd, Janice Whitea, James Grossa, Jennifer L. Matsudaa, Laurent Gapinb, Philippa Marracka,b,e,1, and John W. Kapplera,b,e,1 aDepartment of Biomedical Research, National Jewish Health, Denver, CO 80206; bDepartment of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO 80045; cDivision of Biostatistics and Bioinformatics, National Jewish Health, Denver, CO 80206; dPharmaceutical Sciences, University of Colorado School of Medicine, Aurora, CO 80045; and eHoward Hughes Medical Institute, National Jewish Health, Denver, CO 80206 Contributed by Philippa Marrack, July 6, 2016 (sent for review March 2, 2016; reviewed by Erin J. Adams and Martin Flajnik) The interaction of αβ T-cell antigen receptors (TCRs) with peptides anism termed “negative selection” (13, 14). The remaining T bound to MHC molecules lies at the center of adaptive immunity. cells go on to mature and form the peripheral T-cell repertoire. Whether TCRs have evolved to react with MHC or, instead, pro- The effect of positive and negative thymic selection on limiting cesses in the thymus involving coreceptors and other molecules the T-cell repertoire has made it difficult to test directly whether select MHC-specific TCRs de novo from a random repertoire is a germ-line features of TCRs and MHC molecules have been con- longstanding immunological question. Here, using nuclease-tar- served to promote their interaction. However, some data consistent geted mutagenesis, we address this question in vivo by generating with this notion have accumulated over the past several decades three independent lines of knockin mice with single-amino acid through sequencing, X-ray crystallographic, mutational, and de- mutations of conserved class II MHC amino acids that often are velopmental studies. -

In Murine Resting B Cells Dynamics of MHC Class II-Activating Signals
Dynamics of MHC Class II-Activating Signals in Murine Resting B Cells Toufic O. Nashar and James R. Drake This information is current as J Immunol 2006; 176:827-838; ; of September 26, 2021. doi: 10.4049/jimmunol.176.2.827 http://www.jimmunol.org/content/176/2/827 Downloaded from References This article cites 43 articles, 26 of which you can access for free at: http://www.jimmunol.org/content/176/2/827.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 26, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Dynamics of MHC Class II-Activating Signals in Murine Resting B Cells1 Toufic O. Nashar2 and James R. Drake3 MHC class II (MHC II) proteins are competent signaling molecules on APC. However, little is known about the mechanisms that control generation of their activating signals. -

Cell Differentiation to Plasma Cells B and MHC Class II Gene Expression
ZBTB32 Is an Early Repressor of the CIITA and MHC Class II Gene Expression during B Cell Differentiation to Plasma Cells This information is current as Hye Suk Yoon, Christopher D. Scharer, Parimal Majumder, of September 27, 2021. Carl W. Davis, Royce Butler, Wendy Zinzow-Kramer, Ioanna Skountzou, Dimitrios G. Koutsonanos, Rafi Ahmed and Jeremy M. Boss J Immunol published online 30 July 2012 http://www.jimmunol.org/content/early/2012/07/29/jimmun Downloaded from ol.1103371 Supplementary http://www.jimmunol.org/content/suppl/2012/07/30/jimmunol.110337 Material 1.DC1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published July 30, 2012, doi:10.4049/jimmunol.1103371 The Journal of Immunology ZBTB32 Is an Early Repressor of the CIITA and MHC Class II Gene Expression during B Cell Differentiation to Plasma Cells Hye Suk Yoon,*,1 Christopher D. -

ROLES of Fc RECEPTORS in AUTOIMMUNITY
REVIEWS ROLES OF Fc RECEPTORS IN AUTOIMMUNITY Toshiyuki Takai The receptors for the Fc of immunoglobulins, Fc receptors (FcRs), link the humoral and cellular branches of the immune system, and they have important functions in the activation and down-modulation of immune responses. Balanced signalling through activating and inhibitory FcRs regulates the activity of various cells in the immune system. Recent work in animal models indicates that the development of many human autoimmune diseases might be caused by impairment of the FcR regulatory system. This review provides an overview of the mechanisms of FcR-based immune regulation and describes how autoimmune disease might result from its dysfunction. CENTRAL TOLERANCE Autoimmune diseases are caused by immune responses antibody-mediated autoimmune diseases, I discuss the Refers to the lack of self- to self-antigens in various tissues, and they can be clas- possible involvement of FcRs in T-cell-mediated responsiveness that is established sified broadly as being either antibody mediated or autoimmune diseases. as lymphoid cells develop. It is associated with the deletion of T-cell mediated. Rheumatoid arthritis, for example, autoreactive clones. For T cells, involves joint inflammation and destruction that is Fc-receptor functions this occurs in the thymus. caused by autoimmune antibody responses to Even before the identification of cell-surface receptors unknown synovial antigens. By contrast, patients with for the Fc of antibodies, it was well known that passively PERIPHERAL TOLERANCE type-1 diabetes develop insulitis as a result of T-cell administered antibodies can activate or suppress Refers to the lack of self- 1 responsiveness of mature infiltration and destruction of the pancreatic islets.