Mechanism of Β-Hydrogen Elimination from Square Planar Iridium(I)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Suplementary Information
Supplementary Material (ESI) for Polymer Chemistry This journal is (c) The Royal Society of Chemistry 2011 SUPLEMENTARY INFORMATION Shedding the hydrophobic mantel of polymersomes René P. Brinkhuis, Taco R. Visser, Floris P. J. T. Rutjes, Jan C.M. van Hest* Radboud University Nijmegen,, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands; E-mail: [email protected]; [email protected] Contents: Experimental General note Materials Instrumentation α- methoxy ω-methyl ester poly(ethylene glycol) α- methoxy ω-hydrazide poly(ethylene glycol) α- azido ω-methoxy poly(ethylene glycol) Aldehyde terminated polybutadiene Alkyne terminated polybutadiene Polybutadiene-Hz-poly(ethylene glycol) Polybutadiene-b-poly(ethylene glycol) Vesicle preparation Stability studies Notes and References Figures Figure 1: GPC traces of polymer 1 and its constituents. Figure 2: GPC traces of polymer 2 and its constituents. Figure 3: GPC traces of polymer 3 and its constituents. Figure 4: GPC traces of polymer 4 and its constituents. Supplementary Material (ESI) for Polymer Chemistry This journal is (c) The Royal Society of Chemistry 2011 Experimental section General note: All reactions are described for the lower molecular weight analogue, polyethylene glycol 1000, indicated with an A. The procedures for the polyethylene glycol 2000 analogues, indicated with B, are the same, starting with equimolar amounts. Materials: Sec-butyllithium (ALDRICH 1.4M in hexane), 1,3 butadiene (SIGMA ALDRICH, 99+%), 3- bromo-1-(trimethylsilyl-1-propyne) (ALDRICH, 98%), tetrabutylammonium fluoride (TBAF) (ALDRICH, 1.0M in THF), polyethylene glycol 1000 monomethyl ether (FLUKA), polyethylene glycol 2000 monomethyl ether (FLUKA), methanesulfonyl chloride (MsCl) (JANSSEN CHIMICA, 99%), sodium azide (ACROS ORGANICS, 99% extra pure), sodium hydride (ALDRICH, 60% dispersion in mineral oil), copper bromide (CuBr) (FLUKA, >98%), N,N,N’,N’,N”-penta dimethyldiethylenetriamine (PMDETA) (Aldrich,99%), hydrazine (ALDRICH, 1M in THF) were used as received. -

The Reaction of Sodium Borohydride
THE REACTION OF SODIUM BOROHYDRIDE WITH N-·ACYLANILINES By ROBERT FRA.."\\JK LINDEl"J.ANN Ir Bachelor of Arts Wittenberg College Springfield, Ohio 1952 Submitted to the Faculty of the Graduate School of the Oklahoma Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of MA.STER OF SCIENCE August, 19.54 i THE REACTION OF SODIUM BOROHYDRIDE WITH N-ACYLANILINES Thesis Approved: ~ - Dean of the Graduate School ii l!l Al C! 0~ 11J/~~. 2t"'I'Ii .ACKN01rJLEDGEMENT The author-wishes to express his gratitude to Dr. H. Po Johnston for the invaluable assistance and guidance given. This project was made possible by the Chemistry Department in the form of a Graduate Fellowship and by an unnamed sponsor through the Research Foundation. iii TABLE OF CONTENTS Page · I. .Introduction ...................................... ., o ••••••·· .... 1 II o Historical ... o o • .,, • (I .. ., •.••• G ••••.•••••••• .is •••••••• "' •. o o •••••• o '°. 2 Preparation of Sodium Borohydride ........................... 2 Physical Properties ofSodium Borohydrj.de ................... 3 Chemical Properties of · Sodium Borohydride .............. " •••• 3 III. Experimental. 0 ·• •• ·•. 0 •••• 0 ·• •••• 0. (I •••• 0 0 • 0 Q • .., ••• Q e. 0. 0. 0 D. 0 e 8 Purification of Sodium Borohydride ........................... 8 Reaction of Sodium Borohydride w:i. th Acetanilide ..........._ •• 9 Reaction of Sodium Borohydride w:i. th For.manilide oo •• oe.,. .... 27 Discussion ••• o o. o. o o o o. o ·O fl o • .e. o o ........ o. o. o o • ., o ~. o" o (Io o o o .29 IV. S11ITil11ary •• 0. Cl Ct. 0 0 0. 0. 0 -0 DO O O O C. c,.. 0. 0 4 11) 0 0 0 -0 0 0 0 -0 a O O .0 0 0 .0. -

Sodium Hydride-Based Hydrogen Storage System
Analysis of the Sodium Hydride-based Hydrogen Storage System being developed by PowerBall Technologies, LLC Prepared for The US Department of Energy Office of Power Technologies Hydrogen Program Prepared by J. Philip DiPietro and Edward G. Skolnik, Energetics, Incorporated October 29, 1999 v Analysis of the Sodium Hydride-Based Hydrogen Storage System being developed by PowerBall Technologies, LLC We considered the viability of a system for storing hydrogen on-board a vehicle in the form of plastic-encapsulated sodium hydride (NaH) pellets. Hydrogen is produced when the pellets are cut and immersed in water. The exposed NaH surface reacts with water, releasing hydrogen and forming sodium hydroxide (NaOH) as a byproduct. Later, in an off-board activity, the hydroxide is recycled to hydride via a multi-step regeneration process that relies on methane as both a fuel and a reactant. This is a preliminary analysis that required the development of conceptual designs for several of the process steps and the need to make several assumptions in order to complete the overall systems analysis. The analysis was peer reviewed by several members of their hydrogen community, and their comments were taken into account in the preparation of this document. Executive Summary The reaction of sodium hydride with water to form hydrogen and sodium hydroxide can be utilized onboard a vehicle to deliver hydrogen to an onboard power system. PowerBall Technologies, LLC has developed a novel means of controlling the reaction: they encapsulate small amounts of sodium hydride in plastic balls. These balls are sliced open one at a time onboard the vehicle to deliver hydrogen as needed. -

Metal Hydride Materials for Solid Hydrogen Storage: a Reviewଁ
International Journal of Hydrogen Energy 32 (2007) 1121–1140 www.elsevier.com/locate/ijhydene Review Metal hydride materials for solid hydrogen storage: A reviewଁ Billur Sakintunaa,∗, Farida Lamari-Darkrimb, Michael Hirscherc aGKSS Research Centre, Institute for Materials Research, Max-Planck-Str. 1, Geesthacht D-21502, Germany bLIMHP-CNRS (UPR 1311), Université Paris 13, Avenue J. B. Clément, 93430 Villetaneuse, France cMax-Planck-Institut für Metallforschung, Heisenbergstr. 3, D-70569 Stuttgart, Germany Received 31 July 2006; received in revised form 21 November 2006; accepted 21 November 2006 Available online 16 January 2007 Abstract Hydrogen is an ideal energy carrier which is considered for future transport, such as automotive applications. In this context storage of hydrogen is one of the key challenges in developing hydrogen economy. The relatively advanced storage methods such as high-pressure gas or liquid cannot fulfill future storage goals. Chemical or physically combined storage of hydrogen in other materials has potential advantages over other storage methods. Intensive research has been done on metal hydrides recently for improvement of hydrogenation properties. The present review reports recent developments of metal hydrides on properties including hydrogen-storage capacity, kinetics, cyclic behavior, toxicity, pressure and thermal response. A group of Mg-based hydrides stand as promising candidate for competitive hydrogen storage with reversible hydrogen capacity up to 7.6 wt% for on-board applications. Efforts have been devoted to these materials to decrease their desorption temperature, enhance the kinetics and cycle life. The kinetics has been improved by adding an appropriate catalyst into the system and as well as by ball-milling that introduces defects with improved surface properties. -

Syntheses of Poly(Ethylene Oxide) Macromonomers Carrying Tertiary Amine and Quaternary Ammonium End Groups
Polymer Journal, Vol.35, No. 6, pp 513—518 (2003) Syntheses of Poly(ethylene oxide) Macromonomers Carrying Tertiary Amine and Quaternary Ammonium End Groups † Takamichi SENYO, Yuji ATAGO, Huanan LIANG, Renhua SHEN, and Koichi ITO Department of Materials Science, Toyohashi University of Technology, Tempaku-cho, Toyohashi 441–8580, Japan (Received January 23, 2003; Accepted March 30, 2003) ABSTRACT: p-Vinylbenzyl alcohol, partially alkoxidated with potassium naphthalene, was used successfully to initiate living polymerization of ethylene oxide to afford α-p-vinylbenzyl-ω-hydroxy poly(ethylene oxide) (PEO) macromonomers. The ω-hydroxy end-groups were quantitatively transformed to tertiary amines either by tosylation followed by reaction with potassium 2-dimethylaminoethoxide or by Williamson synthesis with 2-dimethylaminoethyl chloride in the presence of sodium hydride. ω-Quaternary ammonium-ended PEO macromonomers were also quantita- tively obtained by reaction with iodomethane. KEY WORDS Poly(ethylene oxide) / Macromonomers / p-Vinylbenzyl End-Group / Tertiary Amine End-Group / Quaternary Ammonium End-Group / End-Group Transformation / Hetero- telechelics / Poly(ethylene oxide) (PEO) is one of well- ion to afford F– and –OH end-functionalized PEO, known, water-soluble, nonionic polymers, and its F–O[CH2CH2O]n–H, after acidification. In fact, we macromonomers have also been a subject of consid- could readily have α-p-vinylphenylalkyl-ω-hydroxy- erable interest because of their unique amphiphilic ended PEO macromonomers in one step.13 On the other properties as well as their many potential applica- hand, the ω-hydroxy-end may be transformed to intro- tions in various fields, including coatings, cosmetics, duce another functionality, F , to F–O[CH2CH2O]n–F . -

Complex Metal Hydrides for Hydrogen, Thermal and Electrochemical Energy Storage
energies Review Complex Metal Hydrides for Hydrogen, Thermal and Electrochemical Energy Storage Kasper T. Møller 1 ID , Drew Sheppard 1,2, Dorthe B. Ravnsbæk 3 ID , Craig E. Buckley 2, Etsuo Akiba 4,5,6, Hai-Wen Li 1,4,5,7,* and Torben R. Jensen 1,* 1 Interdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, Aarhus University, DK-8000 Aarhus, Denmark; [email protected] (K.T.M.); [email protected] (D.S.) 2 Department of Physics and Astronomy, Fuels and Energy Technology Institute, Curtin University, GPO Box U1987, Perth, WA 6845, Australia; [email protected] 3 Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark; [email protected] 4 International Research Center for Hydrogen Energy, Kyushu University, Fukuoka 819-0395, Japan; [email protected] 5 WPI International Institute for Carbon Neutral Energy Research (WPI-I2CNER), Kyushu University, Fukuoka 819-0395, Japan 6 Department of Mechanical Engineering, Faculty of Engineering, Kyushu University, Fukuoka 819-0395, Japan 7 Kyushu University Platform of Inter/Transdisciplinary Energy Research, Fukuoka 819-0395, Japan * Correspondence: [email protected] (H.-W.L.); [email protected] (T.R.J.) Academic Editor: Haolin Tang Received: 18 September 2017; Accepted: 12 October 2017; Published: 18 October 2017 Abstract: Hydrogen has a very diverse chemistry and reacts with most other elements to form compounds, which have fascinating structures, compositions and properties. Complex metal hydrides are a rapidly expanding class of materials, approaching multi-functionality, in particular within the energy storage field. -

United States Patent Office Patented May 9, 1961
2,983,574 United States Patent Office Patented May 9, 1961 2 moved under vacuum and a substantial amount of solid remained which was purified by crystallization from liq 2,983,574 uid ammonia. The solid was analyzed for sodium, boron and hydrogen and found to be NaBH4. The compound PREPARATION OF SGDIUM BOROHYDRDE. s was further subjected to chemical and X-ray analysis Joseph P. Nigon, Washington, D.C., assignor to Callery which confirmed the fact that it was sodium borohy Chemical Company, Pittsburgh, Pa., a corporation of dride. Pennsylvania Other solvents for NaBH4 may be used as reaction media for this process. Such solvents include poly No Drawing. Filed Sept. 23, 1955, Ser. No. 536,268 O ethylene glycol dimethyl ethers, CH3(OCH)OCHs, and 5 Claims. (C. 23-14) other known solvents for NaBH4 which are not reactive with diborane. The process is operable over a range of temperatures from moderately above room tempera ture i.e., about 20° C., to the boiling point of the solvent This invention relates to the preparation of sodium 15 used. The process is also operable at pressures as low borohydride and more particularly it relates to a direct as 350 p.s.i.g. There is no apparent upper limit of pres method for preparing sodium borohydride by the reac Sc. tion of sodium, diborane and hydrogen under pressure. Although only one embodiment of this invention has There are several known methods for preparing so been described, it will be apparent to those skilled in the dium borohydride among which are the following: (1) 20 art that other variations are possible. -

Technical Data Sheet Page 1 of 3
Technical Data Sheet revision: 24-Jun-09 Product Information: Product: Sodium hydride, 60% dispersion in mineral oil, in soluble bags, in resealable cans Acros code number: 33214-0000 CAS number: 7646-69-7 EINECS number: 231-587-3 TSCA: listed MDL code number: MFCD00003471 Molecular formula: HNa Molecular weight: 23.99 g/mol Typical Properties: Appearance: light grey tacky powder Package: Assay: 57 – 63 % NaH Melting point of NaH: 425 °C (decomposition) Flash point of mineral oil: 165 °C Particle size: 5 – 50 µm Bulk density: approx. 0.60 g/cm3 Solubility: reacts with water, and ethanol Stability: stable in dry air up to 230 °C General Information: Sodium hydride is a very strong base used for condensation reactions like Claisen1,2,3 and Dieckmann4,5 condensation, for C, N, O-alkylation, acylation, Aldol addition, synthesis of sodium alcoholates and sodium borohydride, etc.. Since NaH is sensitive to air and humidity, this product has been packaged in bags* which are soluble in common aprotic organic solvents (see below table), and which makes it easy to bring it directly into a chemical reactor without any complicate handling procedure before. page 1 of 3 Technical Data Sheet revision: 24-Jun-09 * Material: Poly(styrene-co-butadiene); Thickness: ~60 µm Solvent Appearance of the Result solution at 25 °C Cyclohexane clear suitable tert-Butyl methyl ether clear suitable Diethyl ether turbid suitable N,N-Dimethylacetamide turbid suitable N,N-Dimethylformamide turbid suitable Ethylene glycol dimethyl ether clear suitable Heptane bigger parts moderately (turbid at 60 °C) suitable Hexane bigger parts moderately (clear at 60 °C) suitable 2-Methyltetrahydrofuran clear suitable Tetrahydrofuran clear suitable Toluene clear suitable Please notice that Sodium hydride reacts vigorously with water evolving hydrogen (H298 = -132 kJ/mole). -

Supplementary Information
Supplementary Information Direct Synthesis of NaBH4 Nanoparticles from NaOCH3 for Hydrogen Storage Ting Wang† and Kondo-Francois Aguey-Zinsou*,† †MERLin, School of Chemical Engineering, The University of New South Wales, Sydney NSW 2052, Australia, E-mail: f.aguey-unsw.edu.au Table S1. Synthesis methods of NaBH4. Reaction Synthesis and products Yield Ref. Synthesis: React in ether, such as ethyl ether in which NaBH4 is not soluble. 2Na+2B2H6 → NaBH4+NaB3H6 Products: Mixture of NaBH4 and NaB3H6 -- [1,2] with intermediates of empirical composition Na2B2H6 and NaB2H6. Synthesis: Stir approximate proportions of 4Na+2H2+B(OCH3)3 → Na and B(OCH3)3 under H2 pressure at 15% [3,4] NaBH4+3NaOCH3 about 250 °C Synthesis: Disperse Na on finely divided NaCl, add H2 and BCl3, then heat the mixture to 150-170 °C 4Na+2H2+BCl3 → NaBH4+3NaCl Products: NaBH4 is recovered in 75% yield 75% [1] after heating for 3 h. Some diborane and pentaborane are also present as side products. Synthesis: React in ethereal solvents, such as Quantitative 2NaH+B2H6 diglyme 2NaBH4 the glymes, especially diglyme in which [1] yields NaH is more soluble. NaH+B(OCH3)3 → NaBH(OCH3)3 Synthesis: B2H6 react rapidly and Quantitative 2NaBH(OCH3)3+ B2H6 → quantitatively with NaH in the presence of [1] yields 2NaBH4+2B(OCH3)3 B(OCH3)3 90-96% Synthesis: Add methyl borate slowly to an purity excess of a well stirred mass of sodium 4NaH+B(OCH3)3 → NaBH4+3NaOCH3 hydride powder at 225-275 °C. 86-94% Products: NaBH4 may be extracted by liquid yields ammonia or primary amines, such as [1,3, isopropylamine. -

Molecular Transformations Using a Sodium Hydride‑Iodide Composite
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Molecular transformations using a sodium hydride‑iodide composite Chan, Guo Hao 2018 Chan, G. H. (2018). Molecular transformations using a sodium hydride‑iodide composite. Doctoral thesis, Nanyang Technological University, Singapore. https://hdl.handle.net/10356/103312 https://doi.org/10.32657/10220/47383 Downloaded on 04 Oct 2021 04:31:56 SGT MOLECULAR TRANSFORMATIONS USING A SODIUM HYDRIDE-IODIDE COMPOSITE CHAN GUO HAO SCHOOL OF PHYSICAL AND MATHEMATICAL SCIENCES 2018 MOLECULAR TRANSFORMATIONS USING A SODIUM HYDRIDE-IODIDE COMPOSITE ‘ CHAN GUO HAO SCHOOL OF PHYSICAL AND MATHEMATICAL SCIENCES A thesis submitted to the Nanyang Technological University in fulfillment of the requirement for the degree of Doctor of Philosophy 2018 Acknowledgements First and foremost, I would like to express my deepest gratitude to my supervisor, Professor Chiba Shunsuke, whose patient guidance, insightful discussion, and strict training enabled me to complete my PhD course study. His discipline and perspectives toward chemistry and in life will continue to motivate and inspire me. Completing a Ph.D. is not a simple task for me who began with minimal skills and knowledge. I would also like to thank my company supervisor, Jolyon Perkins who always gave great insights to the chemistry process in the industry and encouragement during tough times. I would also like to thank him for giving me the opportunity to participate in some of the company projects for process improvement. I would like to sincerely thank my mentor, Dr. Too Pei Chui, who took her time to teach me the practical skills and the troubleshooting process in the lab. -

Uli1 BOROHYDRIDE with SIMPLE A.ND SUBSTITUTED AMIDES By
THE REACTIONS OF S0D!Uli1 BOROHYDRIDE . WITH SIMPLE A.ND SUBSTITUTED AMIDES By Wo DON BEAVER 11 Bachelor of Arts Bethany-Peniel College Bethanyj Oklahoma 1946 Master of Science Oklahoma Agricultural and Mechanical College Stillwater, Oklahoma 1953 Submitted to the faculty of the Graduate School of the Oklahoma Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY May, 1955 i THE REACTIONS OF SODIUM BOROHYDRIDE WITH SIMPLE AND SUBSTITUTED AMIDES Thesis Approved: z~ M»t4A Dean of the Graduate School 349807 ii ACKNOWLEDGEivlENT The author expresses his thanks to Dr. H.P. Johnston for his gen= erous help and guidance throughout every aspect of this investigationo The contributions of Dr. T. E. Moore and Dr. 0. C. Dermer through helpful criticisms and suggestions are also greatly appreciated. This study was made using the facilities of the Chemistry Depart= ment and was supported by an unnamed sponsor through the Research Found= ation. iii TABLE OF CONTENTS Page I .. INTRODUCTION .. • • 0 • 0 • .. • 0 0 0 O . " 1 IL HISTORICAL • • 0 • 0 • 0 0 .0 0 • 0 o 0 • 0 • • 0 • 0 0 0 2 Preparation of Sodium Borohydride o Ct 0- -0 0 0 0 2 Physical Properties of Sodium Borohydride 0-00000-0 3 Chemical Properties of Sodium Borohydride (I O O O 0 4 Reduction of Amides .. • .. • • • 0(1.P000()0 10 Hydrogenolysis of Amides .. .. • • • • • " • • • • ., • .. o 10 III.. EXPERDIIENT!L I. Reagents and Analytical Procedures 0 0 11 Reagents •••••••••••• 0 o Q 0 11 Analytical Procedures ....... Q00000-00 12 IV. EXPERD11ENTAL IL Reactions . -
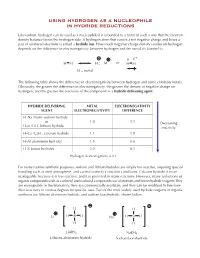
Using Hydrogen As a Nucleophile in Hydride Reductions
USING HYDROGEN AS A NUCLEOPHILE IN HYDRIDE REDUCTIONS Like carbon, hydrogen can be used as a nucleophile if it is bonded to a metal in such a way that the electron density balance favors the hydrogen side. A hydrogen atom that carries a net negative charge and bears a pair of unshared electrons is called a hydride ion. How much negative charge density resides on hydrogen depends on the difference in electronegativity between hydrogen and the metal it’s bonded to. + δ − δ H M H M or H M M = metal The following table shows the difference in electronegativity between hydrogen and some common metals. Obviously, the greater the difference in electronegativity, the greater the density of negative charge on hydrogen, and the greater the reactivity of the compound as a hydride delivering agent. HYDRIDE DELIVERING METAL ELECTRONEGATIVITY AGENT ELECTRONEGATIVITY DIFFERENCE H–Na (NaH) sodium hydride or 1.0 1.1 Decreasing H–Li (LiH) lithium hydride reactivity H–Ca (CaH2) calcium hydride 1.1 1.0 H–Al aluminum hydrides 1.5 0.6 H–B boron hydrides 2.0 0.1 Hydrogen electronegativity is 2.1 For many routine synthetic purposes, sodium and lithium hydrides are simply too reactive, requiring special handling such as inert atmosphere, and careful control of reaction conditions. Calcium hydride is more manageable because it is less reactive, and it is preferred in many reactions. However, many reductions of organic compounds such as carbonyl and carboxyl compounds use aluminum and boron hydride reagents.They are manageable in the laboratory, they are commercially available, and they can be modified to fine-tune their reactivity to various degrees for specific uses.