Blocking Immunoinhibitory Receptor LILRB2 Reprograms Tumor- Associated Myeloid Cells and Promotes Antitumor Immunity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Expression Polarization
Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression This information is current as of September 27, 2021. Fernando O. Martinez, Siamon Gordon, Massimo Locati and Alberto Mantovani J Immunol 2006; 177:7303-7311; ; doi: 10.4049/jimmunol.177.10.7303 http://www.jimmunol.org/content/177/10/7303 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2006/11/03/177.10.7303.DC1 Material http://www.jimmunol.org/ References This article cites 61 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/177/10/7303.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression1 Fernando O. -

Downloaded from GEO
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.17.252007; this version posted November 3, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Oxylipin metabolism is controlled by mitochondrial b-oxidation during bacterial inflammation. Mariya Misheva1, Konstantinos Kotzamanis1*, Luke C Davies1*, Victoria J Tyrrell1, Patricia R S Rodrigues1, Gloria A Benavides2, Christine Hinz1, Robert C Murphy3, Paul Kennedy4, Philip R Taylor1,5, Marcela Rosas1, Simon A Jones1, Sumukh Deshpande1, Robert Andrews1, Magdalena A Czubala1, Mark Gurney1, Maceler Aldrovandi1, Sven W Meckelmann1, Peter Ghazal1, Victor Darley-Usmar2, Daniel White1, and Valerie B O’Donnell1 1Systems Immunity Research Institute and Division of Infection and Immunity, and School of Medicine, Cardiff University, UK, 2Department of Pathology, University of Alabama at Birmingham, Birmingham, AL 35294, USA, 3Department of Pharmacology, University of Colorado Denver, Aurora, CO 80045, USA, 4Cayman Chemical 1180 E Ellsworth Rd, Ann Arbor, MI 48108, United States, 5UK Dementia Research Institute at Cardiff, Cardiff University, UK Address correspondence: Valerie O’Donnell, [email protected] or Daniel White, [email protected], Systems Immunity Research Institute, Cardiff University *Both authors contributed equally to the study 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.17.252007; this version posted November 3, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

BPS Complete Catalog
BPSCATALOG ENZYMES KITS CELL LINES SCREENING SERVICES INNOVATIVE PRODUCTS TO FUEL YOUR EXPERIMENTS Unique, expert portfolio 2017 OUR MISSION: To provide the highest quality life science products and services worldwide in a timely manner to assist in accelerating drug discovery research and development for the treatment of human diseases. INNOVATION WE CONTINUOUSLY STRIVE TO BE FIRST TO MARKET BY PROVIDING EXPERTISE IN EXPRESSING HIGHLY ACTIVE ENZYMES MULTIPLE ASSAY DETECTION FORMATS HUMAN, MURINE AND MONKEY VERSIONS OF RECOMBINANT PROTEINS BIOTINYLATED VERSIONS OF MANY IMMUNE CHECKPOINT RECEPTORS 1ST AND MOST EXTENSIVE PARP ISOZYME PORTFOLIO 1ST AND MOST EXTENSIVE PDE ISOZYME PORTFOLIO 1ST COMPLETE SUITE OF HDAC AND SIRT ENZYMES 1ST AVAILABLE PARP AND TANKYRASE PROFILING SERVICES 1ST COMMERCIALLY AVAILABLE HISTONE DEMETHYLASES LARGEST OFFERING OF METHYLTRANSFERASES LARGEST OFFERING OF DEMETHYLASES OVER 200 PRODUCTS AND SERVICES EXCLUSIVE TO BPS RELIABILITY BPS IS CITED IN PEER-REVIEWED JOURNALS AND PUBLICATIONS AROUND THE WORLD NATURE GENETICS THE JOURNAL OF BIOLOGICAL CHEMISTRY JOURNAL OF CELL SCIENCE ANALYTICAL BIOCHEMISTRY BBRC NATURE CHEMICAL BIOLOGY ACS MEDICINAL CHEMISTRY LETTERS JOURNAL OF NATURAL PRODUCTS CHEMMEDCHEM MOLECULAR CANCER THERAPEUTICS & MANY MORE TABLE OF CONTENTS ACETYLTRANSFERASE 4 APOPTOSIS 4-5 ANTIBODIES 6-9 BIOTINYLATION 10-11 BROMODOMAINS 12-14 CELL BASED ASSAY KITS 15 CELL LINES 16-17 CELL SURFACE RECEPTORS 18-20 CYTOKINE(S) 21-24 DEACETYLASE(S) 25-27 DEMETHYLASE(S) 28-29 HEAT SHOCK PROTEINS 30 IMMUNOTHERAPY -

Antibody List
產品編號 產品名稱 PA569955 1110059E24Rik Polyclonal Antibody PA569956 1110059E24Rik Polyclonal Antibody PA570131 1190002N15Rik Polyclonal Antibody 01-1234-42 123count eBeads Counting Beads MA512242 14.3.3 Pan Monoclonal Antibody (CG15) LFMA0074 14-3-3 beta Monoclonal Antibody (60C10) LFPA0077 14-3-3 beta Polyclonal Antibody PA137002 14-3-3 beta Polyclonal Antibody PA14647 14-3-3 beta Polyclonal Antibody PA515477 14-3-3 beta Polyclonal Antibody PA517425 14-3-3 beta Polyclonal Antibody PA522264 14-3-3 beta Polyclonal Antibody PA529689 14-3-3 beta Polyclonal Antibody MA134561 14-3-3 beta/epsilon/zeta Monoclonal Antibody (3C8) MA125492 14-3-3 beta/zeta Monoclonal Antibody (22-IID8B) MA125665 14-3-3 beta/zeta Monoclonal Antibody (4E2) 702477 14-3-3 delta/zeta Antibody (1H9L19), ABfinity Rabbit Monoclonal 711507 14-3-3 delta/zeta Antibody (1HCLC), ABfinity Rabbit Oligoclonal 702241 14-3-3 epsilon Antibody (5H10L5), ABfinity Rabbit Monoclonal 711273 14-3-3 epsilon Antibody (5HCLC), ABfinity Rabbit Oligoclonal PA517104 14-3-3 epsilon Polyclonal Antibody PA528937 14-3-3 epsilon Polyclonal Antibody PA529773 14-3-3 epsilon Polyclonal Antibody PA575298 14-3-3 eta (Lys81) Polyclonal Antibody MA524792 14-3-3 eta Monoclonal Antibody PA528113 14-3-3 eta Polyclonal Antibody PA529774 14-3-3 eta Polyclonal Antibody PA546811 14-3-3 eta Polyclonal Antibody MA116588 14-3-3 gamma Monoclonal Antibody (HS23) MA116587 14-3-3 gamma Monoclonal Antibody (KC21) PA529690 14-3-3 gamma Polyclonal Antibody PA578233 14-3-3 gamma Polyclonal Antibody 510700 14-3-3 Pan Polyclonal -

Regulation of Tissue Inflammation by 12-Lipoxygenases
biomolecules Review Regulation of Tissue Inflammation by 12-Lipoxygenases Abhishek Kulkarni 1 , Jerry L. Nadler 2, Raghavendra G. Mirmira 1,* and Isabel Casimiro 1,* 1 Department of Medicine, The University of Chicago, Chicago, IL 60637, USA; [email protected] 2 Department of Medicine and Pharmacology, New York Medical College, Valhalla, NY 10595, USA; [email protected] * Correspondence: [email protected] (R.G.M.); [email protected] (I.C.) Abstract: Lipoxygenases (LOXs) are lipid metabolizing enzymes that catalyze the di-oxygenation of polyunsaturated fatty acids to generate active eicosanoid products. 12-lipoxygenases (12-LOXs) primarily oxygenate the 12th carbon of its substrates. Many studies have demonstrated that 12-LOXs and their eicosanoid metabolite 12-hydroxyeicosatetraenoate (12-HETE), have significant pathological implications in inflammatory diseases. Increased level of 12-LOX activity promotes stress (both oxidative and endoplasmic reticulum)-mediated inflammation, leading to damage in these tissues. 12-LOXs are also associated with enhanced cellular migration of immune cells—a characteristic of several metabolic and autoimmune disorders. Genetic depletion or pharmacological inhibition of the enzyme in animal models of various diseases has shown to be protective against disease development and/or progression in animal models in the setting of diabetes, pulmonary, cardiovascular, and metabolic disease, suggesting a translational potential of targeting the enzyme for the treatment of several disorders. In this article, we review the role of 12-LOXs in the pathogenesis of several diseases in which chronic inflammation plays an underlying role. Citation: Kulkarni, A.; Nadler, J.L.; Keywords: 12-lipoxygenases; 12-LOXs; 12/15-lipoxygenase; 12/15-LOX; lipoxygenases; eicosanoids; Mirmira, R.G.; Casimiro, I. -

Original Article Genetic Variants of Aloxs Genes in Polyunsaturated Fatty Acid/Arachidonic Acid Metabolism Associated with Type-2 Diabetes Development
Int J Clin Exp Med 2018;11(12):13797-13805 www.ijcem.com /ISSN:1940-5901/IJCEM0077987 Original Article Genetic variants of ALOXs genes in polyunsaturated fatty acid/arachidonic acid metabolism associated with type-2 diabetes development Jim Jinn-Chyuan Sheu1,3,4,5*, Ying-Ju Lin1,3*, Cherry Yin-Yi Chang2, Shih-Yin Chen1,3, Wen-Ling Liao1,4, Jai-Sing Yang1, Ming-Tsung Lai6, Chih-Mei Chen1, Chun-Cheng Tseng4, Tritium Hwang4, Ping-Ho Chen7, Fuu-Jen Tsai1,3 1Human Genetic Center, 2Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan; 3School of Chinese Medicine, China Medical University, Taichung, Taiwan; 4Institute of Biomedical Sci- ences, National Sun Yat-sen University, Kaohsiung, Taiwan; 5Department of Health and Nutrition Biotechnology, Asia University, Taichung, Taiwan; 6Department of Pathology, Taichung Hospital, Ministry of Health and Welfare, Taichung, Taiwan; 7School of Dentistry, Kaohsiung Medical University, Kaohsiung, Taiwan. *Equal contributors. Received April 16, 2018; Accepted July 24, 2018; Epub December 15, 2018; Published December 30, 2018 Abstract: Poly-unsaturated fatty acids (PUFAs)/arachidonic acids (AAs) and their derived eicosanoids play potent roles in triggering inflammation during obesity and diabetes development. Recent studies have indicated functional roles of ALOX5, ALOX12, ALOX12B, and ALOX15 in the development of insulin resistance and islet β-cell dysfunc- tion. However, the impact of their genetic variants on type 2 diabetes (T2D) development in Asian patients remains unclear. In this study, 1,682 healthy controls and 788 patients with T2D were enrolled for genotyping those four ALOX genes by the TaqMan method. A total of eight Han Chinese-specific SNPs (two SNps for each gene) were selected for this study. -

Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase Is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Spring 2010 Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Michelle Kinder University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Immunology and Infectious Disease Commons Recommended Citation Kinder, Michelle, "Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis" (2010). Publicly Accessible Penn Dissertations. 88. https://repository.upenn.edu/edissertations/88 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/88 For more information, please contact [email protected]. Fatty Acid Metabolism Mediated by 12/15-Lipoxygenase is a Novel Regulator of Hematopoietic Stem Cell Function and Myelopoiesis Abstract Fatty acid metabolism governs critical cellular processes in multiple cell types. The goal of my dissertation was to investigate the intersection between fatty acid metabolism and hematopoiesis. Although fatty acid metabolism has been extensively studied in mature hematopoietic subsets during inflammation, in developing hematopoietic cells the role of fatty acid metabolism, in particular by 12/ 15-Lipoxygenase (12/15-LOX), was unknown. The observation that 12/15-LOX-deficient (Alox15) mice developed a myeloid leukemia instigated my studies since leukemias are often a consequence of dysregulated hematopoiesis. This observation lead to the central hypothesis of this dissertation which is that polyunsaturated fatty acid metabolism mediated by 12/15-LOX participates in hematopoietic development. Using genetic mouse models and in vitro and in vivo cell development assays, I found that 12/15-LOX indeed regulates multiple stages of hematopoiesis including the function of hematopoietic stem cells (HSC) and the differentiation of B cells, T cells, basophils, granulocytes and monocytes. -
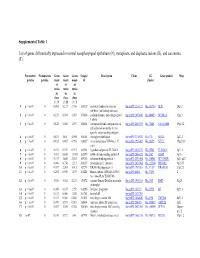
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Functional Characterization of Genetic Enzyme Variations in Human Lipoxygenases
Redox Biology 1 (2013) 566–577 Contents lists available at ScienceDirect Redox Biology journal homepage: www.elsevier.com/locate/redox Functional characterization of genetic enzyme variations in human lipoxygenases Thomas Horn a,n, Kumar Reddy Kakularam b, Monika Anton a, Constanze Richter c, Pallu Reddanna b,d, Hartmut Kuhn a a Institute of Biochemistry, University Medicine Berlin—Charité, Charitéplatz 1, D-10117 Berlin, Germany b Department of Animal Sciences, School of Life Sciences, University of Hyderabad, Gachibowli, Hyderabad 500046, Andhra Pradesh, India c Institute of Nutrition Technology and Nutrition Chemistry, TU Berlin, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany d National Institute of Animal Biotechnology, Hyderabad 500046, Andhra Pradesh, India article info abstract Article history: Mammalian lipoxygenases play a role in normal cell development and differentiation but they have also been Received 28 October 2013 implicated in the pathogenesis of cardiovascular, hyperproliferative and neurodegenerative diseases. As lipid Accepted 1 November 2013 peroxidizing enzymes they are involved in the regulation of cellular redox homeostasis since they produce lipid hydroperoxides, which serve as an efficient source for free radicals. There are various epidemiological Keywords: correlation studies relating naturally occurring variationsinthesixhumanlipoxygenasegenes(SNPsorrare Eicosanoids mutations) to the frequency for various diseases in these individuals, but for most of the described variations no Leukotrienes functional data are available. Employing a combined bioinformatical and enzymological strategy, which Lipoxygenases included structural modeling and experimental site-directed mutagenesis, we systematically explored the Gene polymorphism structural and functional consequences of non-synonymous genetic variations in four different human SNP lipoxygenase genes (ALOX5, ALOX12, ALOX15, and ALOX15B) that have been identified in the human 1000 genome project. -

Analysis of the Impact of Pufas on Placental Gene Expression V2.28 Uploadx
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährungsmedizin Analysis of the Impact of Polyunsaturated Fatty Acids (PUFAs) on Placental Gene Expression Eva-Maria Sedlmeier Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. M. Klingenspor Prüfer der Dissertation: 1. Univ.-Prof. Dr. J. J. Hauner 2. Univ.-Prof. Dr. H. Daniel Die Dissertation wurde am 17.10.2013 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 12.12.2013 angenommen. Table of contents Table of contents Table of contents ....................................................................................................... I Summary .................................................................................................................. VI Zusammenfassung ................................................................................................ VIII 1. Introduction ........................................................................................................ 1 1.1. Fetal programming - a strategy to prevent the obesity epidemic? ...........................................1 1.1.1. The obesity epidemic ........................................................................................................1 1.1.2. Fetal programming -

Acute Lung Injury (ALI) This Information Is Current As of September 28, 2021
Improved Survival and Reduced Vascular Permeability by Eliminating or Blocking 12/15-Lipoxygenase in Mouse Models of Acute Lung Injury (ALI) This information is current as of September 28, 2021. Alexander Zarbock, Matthew R. DiStasi, Emily Smith, John M. Sanders, Gerhard Kronke, Brian L. Harry, Sibylle von Vietinghoff, Konrad Buscher, Jerry L. Nadler and Klaus Ley J Immunol 2009; 183:4715-4722; Prepublished online 14 September 2009; Downloaded from doi: 10.4049/jimmunol.0802592 http://www.jimmunol.org/content/183/7/4715 http://www.jimmunol.org/ References This article cites 59 articles, 15 of which you can access for free at: http://www.jimmunol.org/content/183/7/4715.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 28, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Improved Survival and Reduced Vascular Permeability by Eliminating or Blocking 12/15-Lipoxygenase in Mouse Models of Acute Lung Injury (ALI)1 Alexander Zarbock,2*‡§ Matthew R. -

The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2013 The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach Wüst, Sophia Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-92378 Dissertation Published Version Originally published at: Wüst, Sophia. The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Ap- proach. 2013, University of Zurich, Faculty of Medicine. The Role of the Second 15-Lipoxygenase, ALOX15B, in Atherosclerosis: a Genetic Approach Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr.sc.nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Sophia Julia Annette Wüst von Oberriet-Montlingen SG Promotionskomitee Prof. Dr. Thierry Hennet Prof. Dr. Martin Hersberger (Leitung der Dissertation) Prof. Dr. Arnold von Eckardstein Zürich, 2014 CONTENTS CONTENTS ..............................................................................................1 ABBREVIATIONS.....................................................................................3 SUMMARY................................................................................................6 ZUSAMMENFASSUNG............................................................................8 1. INTRODUCTION ................................................................................10 1.1 Atherosclerosis .................................................................................10