Rheology of Miscible Polymer Blends
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modified PPE-PS Americas
Noryl* resin Modifi ed PPE-PS Americas 2 SABIC Innovative Plastics 1. Introduction Noryl* resin is based on a modified PPO technology developed by SABIC Innovative Plastics. Noryl resin is an extremely versatile material - a miscible blend of PPO resin and polystyrene - and the basic properties may be modified to achieve a variety of characteristics. Contents 1. Introduction 4 2. Applications 6 3. Properties and design 10 3.1 General properties 10 3.2 Mechanical properties 10 3.3 Electrical properties 13 3.4 Flammability 13 3.5 Environmental resistance 14 3.6 Processability 14 3.7 Mold shrinkage 15 4. Processing 16 4.1 Pre-drying 16 4.2 Equipment 16 4.3 Processing conditions 17 4.4 Purging of the barrel 17 4.5 Recycling 17 5. Secondary operations 18 5.1 Welding 18 5.2 Adhesives 19 5.3 Mechanical assembly 20 5.4 Painting 20 5.5 Metalization 21 5.6 Laser marking 22 5.7 Foaming 22 SABIC Innovative Plastics 3 1. Introduction Noryl* modifi ed PPO resin PPE-PS Characteristics Typical characteristics include excellent dimensional stability, low mold shrinkage, low water absorption and very low creep behavior at elevated temperatures. These properties combined with an outstanding hydrolytic stability in hot and cold water, make Noryl resin an excellent potential candidate for fluid engineering, environmental and potable water applications. An outstanding feature of Noryl resin is its retention of tensile and flexural strength, even at elevated temperatures. The gradual reduction in modulus as temperature is increased, is a key advantage of this material. As a result, parts made or molded from Noryl resin may be used with predictable performance over a wide temperature range. -

Poly(Phenylene Ether) Based Amphiphilic Block Copolymers
polymers Review ReviewPoly(phenylene ether) Based Amphiphilic Poly(phenyleneBlock Copolymers ether) Based Amphiphilic Block Copolymers Edward N. Peters EdwardSABIC, Selkirk, N. Peters NY 12158, USA; [email protected]; Tel.: +1-518-475-5458 SABIC, Selkirk, NY 12158, USA; [email protected]; Tel.: +1-518-475-5458 Received: 14 August 2017; Accepted: 5 September 2017; Published: 8 September 2017 Received: 14 August 2017; Accepted: 5 September 2017; Published: 8 September 2017 Abstract: Polyphenylene ether (PPE) telechelic macromonomers are unique hydrophobic polyols Abstract: Polyphenylene ether (PPE) telechelic macromonomers are unique hydrophobic polyols which have been used to prepare amphiphilic block copolymers. Various polymer compositions which have been used to prepare amphiphilic block copolymers. Various polymer compositions have have been synthesized with hydrophilic blocks. Their macromolecular nature affords a range of been synthesized with hydrophilic blocks. Their macromolecular nature affords a range of structures structures including random, alternating, and di- and triblock copolymers. New macromolecular including random, alternating, and di- and triblock copolymers. New macromolecular architectures architectures can offer tailored property profiles for optimum performance. Besides reducing can offer tailored property profiles for optimum performance. Besides reducing moisture uptake and moisture uptake and making the polymer surface more hydrophobic, the PPE hydrophobic segment making the polymer surface more hydrophobic, the PPE hydrophobic segment has good compatibility has good compatibility with polystyrene (polystyrene-philic). In general, the PPE contributes to the with polystyrene (polystyrene-philic). In general, the PPE contributes to the toughness, strength, toughness, strength, and thermal performance. Hydrophilic segments go beyond their affinity for and thermal performance. Hydrophilic segments go beyond their affinity for water. -
Chapter 16-2
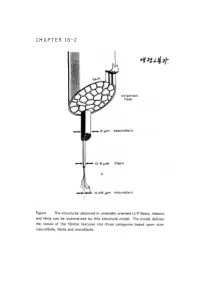
CHAPTER 16-2 ● Polymer Blends (reference) 1. Polymer-polymer Miscibility By Olabisi, Robeson, and Shaw Academic Press(1979) 2. Poplymer Blends, by D.R Paul, Ed, Academic Press (1978). 3. specific Interactions and the Miscibility of Polymer Blends, by Coleman, Graf, and Painter(1990) 4. Polymer Alloys and Blends, Thermodynamics and Rheology, by L.A. Utracki,(1990). ● Blend Preparation 방법: (a) Melt blending- screw extruder를 사용하여 blend하고자 하는 고분자를 용융점 이상에서 extrusion하여 mixing함. (b) Solution blending- mixing하고자 하는 고분자의 cosolvent를 사용하여 solution으로 만든 다음 cast하여 필름(film) 상태 로 blend를 제조함. (ex) polymer membrane(분리막)제조시. 그러나, amorphous polymer와 crystalline polymer를 blend했을때 는 film의 투명성으로부터 사용성을 판별하기는 어렵다. (2)DSC(differential scanning calorimetry): 시차 주사 열분석기를 이용하여 블렌드의 Tg(유리전이 온도)를 측 정함으로서 상용성을 관찰함-가장 많이 쓰이는 방법중 하나임. 즉 (i) a single phase exhibits one Tg. (ex) A polymer : Polycarbonate, Tg=150°C (423 K) B polymer : Polycaprolactane, Tg=-52°C (221 K) Blend (A+B)/50:50인 경우 w w 1 = A + B Tg Blend Tg A Tg B 1 = 0.5 + 0.5 (TgBlend=17.3°C (290.3 K) Tg Blend 423 221 (2) microscopy method (Scanning Electron microscopy). (3) FT-IR (Fourier Transform IR) (4) Ternary solution method (polymer 1- polymer 2 - solvent) - Compatible components from a single, transparent phase in mutual solution, while incompatible polymers exhibit phase seperation if the solution is not extremely dilute. - Equilibrium is relatively easily achived in dilutions and - Blends of immiscible (or partially miscible) materials can be useful so long as no significant desegration occurs while the mixture is being mixed. -

Determination of Polymer Blend Composition, TS-22
Thermal Analysis & Rheology THERMAL SOLUTIONS DETERMINATION OF POLYMER BLEND COMPOSITION PROBLEM SOLUTION The blending of two or more polymers is becoming a Conventional DSC is a technique which measures the total common method for developing new materials for demand- heat flow into and out of a material as a function of ing applications such as impact-resistant parts and temperature and/or time. Hence, conventional DSC packaging films. Since the ultimate properties of blends can measures the sum of all thermal events occurring in the TM be significantly affected by what polymers are present, as material. Modulated DSC , on the other hand, is a new well as by small changes in the blend composition, technique which subjects a material to a linear heating suppliers of these materials are interested in rapid tests method which has a superimposed sinusoidal temperature which provide verification that the correct polymers and oscillation (modulation) resulting in a cyclic heating profile. amount of each polymer are present in the blend. Differen- tial scanning calorimetry (DSC) has proven to be an Deconvolution of the resultant heat flow profile during this effective technique for characterizing blends such as cyclic heating provides not only the "total" heat flow polyethylene/polypropylene where the crystalline melting obtained from conventional DSC, but also separates that endotherms or other transitions (e.g., glass transition) "total" heat flow into its heat capacity-related (reversing) associated with the polymer components are sufficiently and kinetic (nonreversing) components. It is this separa- TM separated to allow identification and/or quantitation. tion aspect which allows MDSC to more completely However, many blends do not exhibit this convenient evaluate polymer blend compositions. -

Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding
polymers Article Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding Maximilian A. Dechet 1,2,3,†, Juan S. Gómez Bonilla 1,2,3,†, Lydia Lanzl 3,4, Dietmar Drummer 3,4, Andreas Bück 1,2,3, Jochen Schmidt 1,2,3 and Wolfgang Peukert 1,2,3,* 1 Institute of Particle Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstraße 4, D-91058 Erlangen, Germany; [email protected] (M.A.D.); [email protected] (J.S.G.B.); [email protected] (A.B.); [email protected] (J.S.) 2 Interdisciplinary Center for Functional Particle Systems, Friedrich-Alexander-Universität Erlangen-Nürnberg, Haberstraße 9a, D-91058 Erlangen, Germany 3 Collaborative Research Center 814—Additive Manufacturing, Am Weichselgarten 9, D-91058 Erlangen, Germany; [email protected] (L.L.); [email protected] (D.D.) 4 Institute of Polymer Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Am Weichselgarten 9, D-91058 Erlangen, Germany * Correspondence: [email protected]; Tel.: +49-9131-85-29400 † The authors contributed equally. Received: 22 November 2018; Accepted: 7 December 2018; Published: 11 December 2018 Abstract: In this study, the feasibility of co-grinding and the subsequent thermal rounding to produce spherical polymer blend particles for selective laser sintering (SLS) is demonstrated for polybutylene terephthalate (PBT) and polycarbonate (PC). The polymers are jointly comminuted in a planetary ball mill, and the obtained product particles are rounded in a heated downer reactor. The size distribution of PBT–PC composite particles is characterized with laser diffraction particle sizing, while the shape and morphology are investigated via scanning electron microscopy (SEM). -

Designing Polymer Blends Using Neural Networks, Genetic
Designing Polymer Blends Using Neural Networks, Genetic Algorithms, and Markov Chains N. K. Roy1,2, W. D. Potter1, D. P. Landau2 1Department of Computer Science University of Georgia, Athens, GA 30602 2Center for Simulational Physics University of Georgia, Athens, GA 30602 ABSTRACT In this paper we present a new technique to simulate polymer blends that overcomes the shortcomings in polymer system modeling. This method has an inherent advantage in that the vast existing information on polymer systems forms a critical part in the design process. The stages in the design begin with selecting potential candidates for blending using Neural Networks. Generally the parent polymers of the blend need to have certain properties and if the blend is miscible then it will reflect the properties of the parents. Once this step is finished the entire problem is encoded into a genetic algorithm using various models as fitness functions. We select the lattice fluid model of Sanchez and Lacombe1, which allows for a compressible lattice. After reaching a steady-state with the genetic algorithm we transform the now stochastic problem that satisfies detailed balance and the condition of ergodicity to a Markov Chain of states. This is done by first creating a transition matrix, and then using it on the incidence vector obtained from the final populations of the genetic algorithm. The resulting vector is converted back into a 1 population of individuals that can be searched to find the individuals with the best fitness values. A high degree of convergence not seen using the genetic algorithm alone is obtained. We check this method with known systems that are miscible and then use it to predict miscibility on several unknown systems. -

Phase Behavior of Polymer Solutions and Blends Piotr Knychała,† Ksenia Timachova,‡ Michał Banaszak,*,∥,⊥ and Nitash P
Perspective pubs.acs.org/Macromolecules 50th Anniversary Perspective: Phase Behavior of Polymer Solutions and Blends Piotr Knychała,† Ksenia Timachova,‡ Michał Banaszak,*,∥,⊥ and Nitash P. Balsara*,‡,# † Faculty of Polytechnic, The President Stanislaw Wojciechowski State University of Applied Sciences, Kalisz, Poland ‡ Department of Chemical and Biomolecular Engineering, University of California, Berkeley, Berkeley, California 94720, United States ∥ ⊥ Faculty of Physics and NanoBioMedical Centre, Adam Mickiewicz University, ul. Umultowska 85, 61-614 Poznan, Poland # Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States *S Supporting Information ABSTRACT: We summarize our knowledge of the phase behavior of polymer solutions and blends using a unified approach. We begin with a derivation of the Flory−Huggins expression for the Gibbs free energy of mixing two chemically dissimilar polymers. The Gibbs free energy of mixing of polymer solutions is obtained as a special case. These expressions are used to interpret observed phase behavior of polymer solutions and blends. Temperature- and pressure-dependent phase diagrams are used to determine the Flory−Huggins interaction parameter, χ. We also discuss an alternative approach for measuring χ due to de Gennes, who showed that neutron scattering from concentration fluctuations in one-phase systems was a sensitive function of χ. In most cases, the agreement between experimental data and the standard Flory−Huggins−de Gennes approach is qualitative. We conclude by summarizing advanced theories that have been proposed to address the limitations of the standard approach. In spite of considerable effort, there is no consensus on the reasons for departure between the standard theories and experiments. ■ INTRODUCTION mean-field theory of de Gennes and their effect on phase 4,5 Our understanding of the thermodynamic underpinnings of the behavior. -

Effects of Polystyrene-Modified
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repository@USM EFFECTS OF POLYSTYRENE-MODIFIED NATURAL RUBBER ON THE PROPERTIES OF POLYPROPYLENE/POLYSTYRENE BLENDS by SIA CHOON SIONG Thesis submitted in fulfilment of the requirements for the degree of Master of Science DECEMBER 2008 ACKNOWLEDGEMENTS First and foremost, I would like to extend my sincerest gratitude to my parents and siblings for their love, unconditional support and plenty of encouragement to me although we are financially-challenged during the first stages of my journey. It’s so happen that I was very persistent in pursuing my dream that I had burdened all of you both mentally and financially. Subsequently, this has have become OUR dream and without you, it will be mission impossible for me to achieve current success. My deepest love and appreciation for all of you that stood by me through all these challenging times. I am totally indebted to my direct supervisor Prof. Azanam Shah Hashim for his constant dedication, assistance, advice and guidance that propelled me to a more endowed researcher armed with a balanced skill to carry out my research with great perseverance and dedication. It’s your “First Time Right” mentally that has been our motto to ensure this research a smooth sailing journey. We enjoyed each others’ company during the times of research and not forgetting the lunches that we occasionally have during break time. Truly a memorable time that I will always cherish in my heart. Special acknowledgement is accorded to my fellow ‘brothers’ Yap Gay Suu and Chin Kian Meng for their support and inputs that made this research a joyful and pleasant journey. -

Commercial Polymer Blends
CHAPTER 15 COMMERCIAL POLYMER BLENDS M. K. AKKAPEDDI Honeywell Inc., EAS R & T, Morristown, USA 15.1 Abstract In this chapter, an overview of the commercially important blends is presented with a particular emphasis on the rationale for their commercial development, the compatibilization principles, their key mechanical properties and their current applica- tions and markets. To facilitate the discussion, the commercial polymer blends have been classifi ed into twelve major groups depending on the type of the resin family they are based on, viz. (i) polyolefi n, (ii) styrenic, (iii) vinyl, (iv) acrylic, (v) elastomeric, (vi) polyamide, (vii) polycarbonate, (viii) poly(oxymethylene), (ix) polyphenyleneether, (x) thermoplastic polyester, (xi) specialty polymers, and (xii) thermoset blends. Within each major category, the individual polymer blends of industrial signifi cance have been described with relevant data. Since the discussion is limited only to those blends that are actually produced and used on a commercial scale, the relevant cost and performance factors that contribute to the commercial viability and success of various types of blends have been outlined. In comparing the different blends, the specifi c advantages of each type, as well as any potential overlap in performance with other type of blends have also been discussed. The fundamental advantage of polymer blends viz. their ability to combine cost-effectively the unique features of individual resins, is particularly illustrated in the discussion of crystalline/amorphous polymer blends, such as the polyamide and the polyester blends. Key to the success of many commercial blends, however, is in the selection of intrinsically complementing systems or in the development of effective compatibilization method. -

Historical Perspective of Advances in the Science and Technology of Polymer Blends
Polymers 2014, 6, 1251-1265; doi:10.3390/polym6051251 OPEN ACCESS polymers ISSN 2073-4360 www.mdpi.com/journal/polymers Review Historical Perspective of Advances in the Science and Technology of Polymer Blends Lloyd Robeson Department of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA; E-Mail: [email protected]; Tel.: +1-610-481-0117 Received: 21 March 2014; in revised form: 14 April 2014 / Accepted: 25 April 2014 / Published: 30 April 2014 Abstract: This paper will review the important developments in the field of polymer blends. The subject of polymer blends has been one of the most prolific areas in polymer science and technology in the past five decades judging from publications and patents on the subject. Although a continuing important subject, the peak intensity occurred in the 1970s and 1980s. The author has been active in this area for five decades and this paper is a recollection of some of the important milestones/breakthroughs in the field. The discussion will cover the development of the theory relevant to polymer blends, experimental methods, approaches to achieve compatibility in immiscible/incompatible blends, the nature of phase separation and commercial activity. Keywords: polymer blends; polymer miscibility; polymer blend compatibilization; historical review of polymer blends 1. Introduction The area of polymer blends has been a major topic of polymer research and development for almost five decades. Although noted to be of interest much earlier, the academic and industrial effort in polymer blends exponentially increased starting in the late 1960s. Although various reasons were responsible for this increased interest, one significant factor was the emergence of a major engineering polymer blend based on polystyrene (specifically impact polystyrene) and poly(2,6-dimethyl-1,4-phenylene oxide) (PPO). -

Interpenetrating Polymer Network in Drug Delivery Formulations: Revisited
Online - 2455-3891 Vol 12, Issue 6, 2019 Print - 0974-2441 Review Article INTERPENETRATING POLYMER NETWORK IN DRUG DELIVERY FORMULATIONS: REVISITED SUBHASISH PRAMANIK*, PRITHVIRAJ CHAKRABORTY Department of Pharmaceutics, Himalayan Pharmacy Institute, Majhitar, Sikkim, India. Email: [email protected] Received: 18 March 2019, Revised and Accepted: 22 April 2019 ABSTRACT Interpenetrating polymer network has gained a lot of interest in drug delivery system due to its ease of modification during its synthesis and development state, which evolved novel physicochemical and mechanical properties within the formulation. In this work, a detailed revision was done which includes its methods of preparations, factors that influence their properties, basic characterization parameters, and use in drug delivery systems along with patented formulations. Keywords: Interpenetrating polymer network, Characterization, Semi-interpenetrating polymer network. © 2019 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons. org/licenses/by/4. 0/) DOI: http://dx.doi.org/10.22159/ajpcr.2019.v12i6.33117 INTRODUCTION agent, the IPN properties can be enhanced which precisely include porosity, elasticity, and degree of swelling and responsive behavior [10]. In the modern science of drug discovery, the polymer is widely used and valuable excipients in the different pharmaceutical formulation. KINDS OF IPN It also has a high performance in the parenteral area, controlled drug release, and drug targeting to specific organs. Now in recent years, IPN can be ordered in many different ways (Fig. 2). polymer mixture or blending is used to improve the polymer properties to subside the poor biological properties or progress in mechanical Based on chemical bonding strength of the polymer [1]. -

Polymer Blend and Composite
Polymer blend and Composite (Lecture 3 ) Dr.Vandana K. Nair Polymer Blends • Combination of two or more polymers or copolymers. • 3 types: • Immiscible (heterogeneous) polymer blends: • example: high-impact polystyrene 2. Compatible polymer blends • Blends that miscible in a certain useful range of composition and temperature, but immiscible in others. • Example: PC/ABS blends (Poly carbonate-Acrylonitrile butadiene styrene) • 3. Miscible polymer blends ( homogeneous) : Single phase structure. • Example: PET-PBT ( Poly ethylene terephthalate- Polybutylene terephthalate Properties • Appear as separate phases, when viewed under a microscope. • Between different polymer chains, only vander waals forces or hydrogen bonding exists. • The properties are closely related to the properties of the individual components. • Sensitive component of a blend may be protected from degradation by blending. • Eg. PMMA is degraded by gamma-radiation. But its blend with styrene-acrylonitrile copolymer reduces the rate of degradation. • Blending increases the properties like flame retardence, abrasive resistance etc. Application • Polycarbonate – Acrylonitrile butadiene styrene (PC- ABS) blend is used for making machine parts. • Nylon 6-PC blend is used for making sport equipments and transport containers. • Polydimethylphenylene –polystyrene ( PDP-PS) blend possesses low water absorption, resistance to hydrolysis. Its used in electrical industries, radio and TV parts and automobile parts. • ABS plastics( Acrylonitrile styrene with Butadiene styrene rubber). Posses high impact strength and high mouldability. ABS products are used in automobile industry for making panels, door bands, door covers etc. COMPOSITES • Material made from two or more constituent materials with significantly different physical or chemical properties that, when combined, produce a material with characteristics different from the individual components.