Composition Dependence of the Flory-Huggins Interaction Parameter in Polymer Blends: Structural and Thermodynamic Calculations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Properties of Two-Dimensional Polymers
Macromolecules 1982,15, 549-553 549 The squared frequencies uA2are obtained from the ei- (4) Flory, P. J. "Statistical Mechanics of Chain Molecules"; In- genvalues Of this matrix described in the text* terscience: New York, 1969; p 315. (5) Volkenstein,M. V. &Configurational Statistics of polymeric for N = 3 and 4 will be found in ref 7. Chains" (translated from the Russian edition by S. N. Ti- masheff and M. J. Timasheff);Interscience: New York, 1963; References and Notes p 450. (1) Weiner, J. H.; Perchak, D. Macromolecules 1981, 14, 1590. (6) Frenkel, J. "Kinetic Theory of Liquids"; Oxford University (2) Weiner, J. H., Macromolecules, preceding paper in this issue. Press: London, 1946, Dover reprint, 1955; pp 474-476. (3) G6, N.; Scheraga, H. A. Macromolecules 1976, 9, 535. (7) Perchak, D. Doctoral Dissertation, Brown University, 1981. Properties of Two-Dimensional Polymers Jan Tobochnik and Itzhak Webman* Department of Mathematics, Rutgers, The State University, New Brunswick, New Jersey 08903 Joel L. Lebowitzt Institute for Advanced Study, Princeton, New Jersey 08540 M. H. Kalos Courant Institute of Mathematical Sciences, New York university, New York, New York 10012. Received August 3, 1981 ABSTRACT: We have performed a series of Monte Carlo simulations for a two-dimensional polymer chain with monomers interacting via a Lennard-Jones potential. An analysis of a mean field theory, based on approximating the free energy as the sum of an elastic part and a fluid part, shows that in two dimensions there is a sharp collapse transition, at T = 0, but that there is no ideal or quasi-ideal behavior at the transition as there is in three dimensions. -

Modified PPE-PS Americas
Noryl* resin Modifi ed PPE-PS Americas 2 SABIC Innovative Plastics 1. Introduction Noryl* resin is based on a modified PPO technology developed by SABIC Innovative Plastics. Noryl resin is an extremely versatile material - a miscible blend of PPO resin and polystyrene - and the basic properties may be modified to achieve a variety of characteristics. Contents 1. Introduction 4 2. Applications 6 3. Properties and design 10 3.1 General properties 10 3.2 Mechanical properties 10 3.3 Electrical properties 13 3.4 Flammability 13 3.5 Environmental resistance 14 3.6 Processability 14 3.7 Mold shrinkage 15 4. Processing 16 4.1 Pre-drying 16 4.2 Equipment 16 4.3 Processing conditions 17 4.4 Purging of the barrel 17 4.5 Recycling 17 5. Secondary operations 18 5.1 Welding 18 5.2 Adhesives 19 5.3 Mechanical assembly 20 5.4 Painting 20 5.5 Metalization 21 5.6 Laser marking 22 5.7 Foaming 22 SABIC Innovative Plastics 3 1. Introduction Noryl* modifi ed PPO resin PPE-PS Characteristics Typical characteristics include excellent dimensional stability, low mold shrinkage, low water absorption and very low creep behavior at elevated temperatures. These properties combined with an outstanding hydrolytic stability in hot and cold water, make Noryl resin an excellent potential candidate for fluid engineering, environmental and potable water applications. An outstanding feature of Noryl resin is its retention of tensile and flexural strength, even at elevated temperatures. The gradual reduction in modulus as temperature is increased, is a key advantage of this material. As a result, parts made or molded from Noryl resin may be used with predictable performance over a wide temperature range. -

Poly(Phenylene Ether) Based Amphiphilic Block Copolymers
polymers Review ReviewPoly(phenylene ether) Based Amphiphilic Poly(phenyleneBlock Copolymers ether) Based Amphiphilic Block Copolymers Edward N. Peters EdwardSABIC, Selkirk, N. Peters NY 12158, USA; [email protected]; Tel.: +1-518-475-5458 SABIC, Selkirk, NY 12158, USA; [email protected]; Tel.: +1-518-475-5458 Received: 14 August 2017; Accepted: 5 September 2017; Published: 8 September 2017 Received: 14 August 2017; Accepted: 5 September 2017; Published: 8 September 2017 Abstract: Polyphenylene ether (PPE) telechelic macromonomers are unique hydrophobic polyols Abstract: Polyphenylene ether (PPE) telechelic macromonomers are unique hydrophobic polyols which have been used to prepare amphiphilic block copolymers. Various polymer compositions which have been used to prepare amphiphilic block copolymers. Various polymer compositions have have been synthesized with hydrophilic blocks. Their macromolecular nature affords a range of been synthesized with hydrophilic blocks. Their macromolecular nature affords a range of structures structures including random, alternating, and di- and triblock copolymers. New macromolecular including random, alternating, and di- and triblock copolymers. New macromolecular architectures architectures can offer tailored property profiles for optimum performance. Besides reducing can offer tailored property profiles for optimum performance. Besides reducing moisture uptake and moisture uptake and making the polymer surface more hydrophobic, the PPE hydrophobic segment making the polymer surface more hydrophobic, the PPE hydrophobic segment has good compatibility has good compatibility with polystyrene (polystyrene-philic). In general, the PPE contributes to the with polystyrene (polystyrene-philic). In general, the PPE contributes to the toughness, strength, toughness, strength, and thermal performance. Hydrophilic segments go beyond their affinity for and thermal performance. Hydrophilic segments go beyond their affinity for water. -
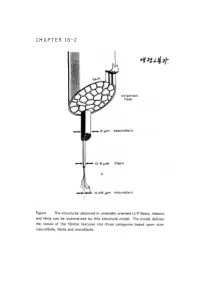
Chapter 16-2
CHAPTER 16-2 ● Polymer Blends (reference) 1. Polymer-polymer Miscibility By Olabisi, Robeson, and Shaw Academic Press(1979) 2. Poplymer Blends, by D.R Paul, Ed, Academic Press (1978). 3. specific Interactions and the Miscibility of Polymer Blends, by Coleman, Graf, and Painter(1990) 4. Polymer Alloys and Blends, Thermodynamics and Rheology, by L.A. Utracki,(1990). ● Blend Preparation 방법: (a) Melt blending- screw extruder를 사용하여 blend하고자 하는 고분자를 용융점 이상에서 extrusion하여 mixing함. (b) Solution blending- mixing하고자 하는 고분자의 cosolvent를 사용하여 solution으로 만든 다음 cast하여 필름(film) 상태 로 blend를 제조함. (ex) polymer membrane(분리막)제조시. 그러나, amorphous polymer와 crystalline polymer를 blend했을때 는 film의 투명성으로부터 사용성을 판별하기는 어렵다. (2)DSC(differential scanning calorimetry): 시차 주사 열분석기를 이용하여 블렌드의 Tg(유리전이 온도)를 측 정함으로서 상용성을 관찰함-가장 많이 쓰이는 방법중 하나임. 즉 (i) a single phase exhibits one Tg. (ex) A polymer : Polycarbonate, Tg=150°C (423 K) B polymer : Polycaprolactane, Tg=-52°C (221 K) Blend (A+B)/50:50인 경우 w w 1 = A + B Tg Blend Tg A Tg B 1 = 0.5 + 0.5 (TgBlend=17.3°C (290.3 K) Tg Blend 423 221 (2) microscopy method (Scanning Electron microscopy). (3) FT-IR (Fourier Transform IR) (4) Ternary solution method (polymer 1- polymer 2 - solvent) - Compatible components from a single, transparent phase in mutual solution, while incompatible polymers exhibit phase seperation if the solution is not extremely dilute. - Equilibrium is relatively easily achived in dilutions and - Blends of immiscible (or partially miscible) materials can be useful so long as no significant desegration occurs while the mixture is being mixed. -

Determination of Polymer Blend Composition, TS-22
Thermal Analysis & Rheology THERMAL SOLUTIONS DETERMINATION OF POLYMER BLEND COMPOSITION PROBLEM SOLUTION The blending of two or more polymers is becoming a Conventional DSC is a technique which measures the total common method for developing new materials for demand- heat flow into and out of a material as a function of ing applications such as impact-resistant parts and temperature and/or time. Hence, conventional DSC packaging films. Since the ultimate properties of blends can measures the sum of all thermal events occurring in the TM be significantly affected by what polymers are present, as material. Modulated DSC , on the other hand, is a new well as by small changes in the blend composition, technique which subjects a material to a linear heating suppliers of these materials are interested in rapid tests method which has a superimposed sinusoidal temperature which provide verification that the correct polymers and oscillation (modulation) resulting in a cyclic heating profile. amount of each polymer are present in the blend. Differen- tial scanning calorimetry (DSC) has proven to be an Deconvolution of the resultant heat flow profile during this effective technique for characterizing blends such as cyclic heating provides not only the "total" heat flow polyethylene/polypropylene where the crystalline melting obtained from conventional DSC, but also separates that endotherms or other transitions (e.g., glass transition) "total" heat flow into its heat capacity-related (reversing) associated with the polymer components are sufficiently and kinetic (nonreversing) components. It is this separa- TM separated to allow identification and/or quantitation. tion aspect which allows MDSC to more completely However, many blends do not exhibit this convenient evaluate polymer blend compositions. -

Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding
polymers Article Spherical Polybutylene Terephthalate (PBT)—Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding Maximilian A. Dechet 1,2,3,†, Juan S. Gómez Bonilla 1,2,3,†, Lydia Lanzl 3,4, Dietmar Drummer 3,4, Andreas Bück 1,2,3, Jochen Schmidt 1,2,3 and Wolfgang Peukert 1,2,3,* 1 Institute of Particle Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstraße 4, D-91058 Erlangen, Germany; [email protected] (M.A.D.); [email protected] (J.S.G.B.); [email protected] (A.B.); [email protected] (J.S.) 2 Interdisciplinary Center for Functional Particle Systems, Friedrich-Alexander-Universität Erlangen-Nürnberg, Haberstraße 9a, D-91058 Erlangen, Germany 3 Collaborative Research Center 814—Additive Manufacturing, Am Weichselgarten 9, D-91058 Erlangen, Germany; [email protected] (L.L.); [email protected] (D.D.) 4 Institute of Polymer Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Am Weichselgarten 9, D-91058 Erlangen, Germany * Correspondence: [email protected]; Tel.: +49-9131-85-29400 † The authors contributed equally. Received: 22 November 2018; Accepted: 7 December 2018; Published: 11 December 2018 Abstract: In this study, the feasibility of co-grinding and the subsequent thermal rounding to produce spherical polymer blend particles for selective laser sintering (SLS) is demonstrated for polybutylene terephthalate (PBT) and polycarbonate (PC). The polymers are jointly comminuted in a planetary ball mill, and the obtained product particles are rounded in a heated downer reactor. The size distribution of PBT–PC composite particles is characterized with laser diffraction particle sizing, while the shape and morphology are investigated via scanning electron microscopy (SEM). -

Designing Polymer Blends Using Neural Networks, Genetic
Designing Polymer Blends Using Neural Networks, Genetic Algorithms, and Markov Chains N. K. Roy1,2, W. D. Potter1, D. P. Landau2 1Department of Computer Science University of Georgia, Athens, GA 30602 2Center for Simulational Physics University of Georgia, Athens, GA 30602 ABSTRACT In this paper we present a new technique to simulate polymer blends that overcomes the shortcomings in polymer system modeling. This method has an inherent advantage in that the vast existing information on polymer systems forms a critical part in the design process. The stages in the design begin with selecting potential candidates for blending using Neural Networks. Generally the parent polymers of the blend need to have certain properties and if the blend is miscible then it will reflect the properties of the parents. Once this step is finished the entire problem is encoded into a genetic algorithm using various models as fitness functions. We select the lattice fluid model of Sanchez and Lacombe1, which allows for a compressible lattice. After reaching a steady-state with the genetic algorithm we transform the now stochastic problem that satisfies detailed balance and the condition of ergodicity to a Markov Chain of states. This is done by first creating a transition matrix, and then using it on the incidence vector obtained from the final populations of the genetic algorithm. The resulting vector is converted back into a 1 population of individuals that can be searched to find the individuals with the best fitness values. A high degree of convergence not seen using the genetic algorithm alone is obtained. We check this method with known systems that are miscible and then use it to predict miscibility on several unknown systems. -

Development of the Semi-Empirical Equation of State for Square-Well Chain Fluid Based on the Statistical Associating Fluid Theory (SAFT)
Korean J. Chem. Eng., 17(1), 52-57 (2000) Development of the Semi-empirical Equation of State for Square-well Chain Fluid Based on the Statistical Associating Fluid Theory (SAFT) Min Sun Yeom, Jaeeon Chang and Hwayong Kim† School of Chemical Engineering, Seoul National University, Shinlim-dong, Kwanak-ku, Seoul 151-742, Korea (Received 25 August 1999 • accepted 29 October 1999) Abstract−A semi-empirical equation of state for the freely jointed square-well chain fluid is developed. This equation of state is based on Wertheim's thermodynamic perturbation theory (TPT) and the statistical associating fluid theory (SAFT). The compressibility factor and radial distribution function of square-well monomer are ob- tained from Monte Carlo simulations. These results are correlated using density expansion. In developing the equa- tion of state the exact analytical expressions are adopted for the second and third virial coefficients for the com- pressibility factor and the first two terms of the radial distribution function, while the higher order coefficients are determined from regression using the simulation data. In the limit of infinite temperature, the present equation of state and the expression for the radial distribution function are represented by the Carnahan-Starling equation of state. This semi-empirical equation of state gives at least comparable accuracy with other empirical equation of state for the square-well monomer fluid. With the new SAFT equation of state from the accurate expressions for the mo- nomer reference and covalent terms, we compare the prediction of the equation of state to the simulation results for the compressibility factor and radial distribution function of the square-well monomer and chain fluids. -

Phase Behavior of Polymer Solutions and Blends Piotr Knychała,† Ksenia Timachova,‡ Michał Banaszak,*,∥,⊥ and Nitash P
Perspective pubs.acs.org/Macromolecules 50th Anniversary Perspective: Phase Behavior of Polymer Solutions and Blends Piotr Knychała,† Ksenia Timachova,‡ Michał Banaszak,*,∥,⊥ and Nitash P. Balsara*,‡,# † Faculty of Polytechnic, The President Stanislaw Wojciechowski State University of Applied Sciences, Kalisz, Poland ‡ Department of Chemical and Biomolecular Engineering, University of California, Berkeley, Berkeley, California 94720, United States ∥ ⊥ Faculty of Physics and NanoBioMedical Centre, Adam Mickiewicz University, ul. Umultowska 85, 61-614 Poznan, Poland # Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States *S Supporting Information ABSTRACT: We summarize our knowledge of the phase behavior of polymer solutions and blends using a unified approach. We begin with a derivation of the Flory−Huggins expression for the Gibbs free energy of mixing two chemically dissimilar polymers. The Gibbs free energy of mixing of polymer solutions is obtained as a special case. These expressions are used to interpret observed phase behavior of polymer solutions and blends. Temperature- and pressure-dependent phase diagrams are used to determine the Flory−Huggins interaction parameter, χ. We also discuss an alternative approach for measuring χ due to de Gennes, who showed that neutron scattering from concentration fluctuations in one-phase systems was a sensitive function of χ. In most cases, the agreement between experimental data and the standard Flory−Huggins−de Gennes approach is qualitative. We conclude by summarizing advanced theories that have been proposed to address the limitations of the standard approach. In spite of considerable effort, there is no consensus on the reasons for departure between the standard theories and experiments. ■ INTRODUCTION mean-field theory of de Gennes and their effect on phase 4,5 Our understanding of the thermodynamic underpinnings of the behavior. -

High Elasticity of Polymer Networks. Polymer Networks
High Elasticity of Polymer Networks. Polymer Networks. Polymer network consists of long polymer chains which are crosslinked with each other to form a giant three-dimensional macromolecule. All polymer networks except those who are in the glassy or partially crystalline states exhibit the property of high elasticity, i.e., the ability to undergo large reversible deformations at relatively small applied stress. High elasticity is the most specific property of polymer materials, it is connected with the most fundamental features of ideal chains considered in the previous lecture. In everyday life, highly elastic polymer materials are called rubbers. Molecular Nature of the High Elasticity. Elastic response of the crystalline solids is due to the change of the equilibrium interatomic distances under stress and therefore, the change in the internal energy of the crystal. Elasticity of rubbers is composed from the elastic responses of the chains crosslinked in the network sample. External stress changes the equilibrium end-to-end distance of a chain, and it thus adopts a less probable conformation. Therefore, the elasticity of rubbers is of purely entropic nature. Typical Stress-Strain Curves. for steel for rubbers А - the upper limit for stress-strain linearity, В - the upper limit for the reversibility of deformations, С - the fracture point. - the typical values of deformations ∆ l l are much larger for rubber; - the typical values of strain σ are much larger for steel; - the typical values of the Young modulus is enormously larger for steel than for rubber ( E 1 0 1 1 Pa and 1 0 5 − 1 0 6 Pa, respectively); - for steel linearity and reversibility are lost practically simultaneously, while for rubbers there is a very wide region of nonlinear reversible deformations; - for steel there is a wide region of plastic deformations (between points B and C) which is all but absent for rubbers. -

Introduction to Polymer Physics Lecture 1 Boulder Summer School July – August 3, 2012
Introduction to polymer physics Lecture 1 Boulder Summer School July – August 3, 2012 A.Grosberg New York University Lecture 1: Ideal chains • Course outline: • This lecture outline: •Ideal chains (Grosberg) • Polymers and their •Real chains (Rubinstein) uses • Solutions (Rubinstein) •Scales •Methods (Grosberg) • Architecture • Closely connected: •Polymer size and interactions (Pincus), polyelectrolytes fractality (Rubinstein) , networks • Entropic elasticity (Rabin) , biopolymers •Elasticity at high (Grosberg), semiflexible forces polymers (MacKintosh) • Limits of ideal chain Polymer molecule is a chain: Examples: polyethylene (a), polysterene •Polymericfrom Greek poly- (b), polyvinyl chloride (c)… merēs having many parts; First Known Use: 1866 (Merriam-Webster); • Polymer molecule consists of many elementary units, called monomers; • Monomers – structural units connected by covalent bonds to form polymer; • N number of monomers in a …and DNA polymer, degree of polymerization; •M=N*mmonomer molecular mass. Another view: Scales: •kBT=4.1 pN*nm at • Monomer size b~Å; room temperature • Monomer mass m – (240C) from 14 to ca 1000; • Breaking covalent • Polymerization bond: ~10000 K; degree N~10 to 109; bondsbonds areare NOTNOT inin • Contour length equilibrium.equilibrium. L~10 nm to 1 m. • “Bending” and non- covalent bonds compete with kBT Polymers in materials science (e.g., alkane hydrocarbons –(CH2)-) # C 1-5 6-15 16-25 20-50 1000 or atoms more @ 25oC Gas Low Very Soft solid Tough and 1 viscosity viscous solid atm liquid liquid Uses Gaseous