New Mutation, P575L, in the GUCY2D Gene in a Family with Autosomal Dominant Progressive Cone Degeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GUCY2D Cone-Rod Dystrophy-6 Is a ‘Phototransduction Disease’ Triggered by Abnormal Calcium Feedback on Retinal Membrane Guanylyl Cyclase 1
This Accepted Manuscript has not been copyedited and formatted. The final version may differ from this version. Research Articles: Neurobiology of Disease GUCY2D Cone-Rod Dystrophy-6 is a ‘Phototransduction Disease’ Triggered by Abnormal Calcium Feedback on Retinal Membrane Guanylyl Cyclase 1 Shinya Sato1, Igor V. Peshenko2, Elena V. Olshevskaya2, Vladimir J. Kefalov1 and Alexander M. Dizhoor2 1Department of Ophthalmology and Visual Sciences, Washington University in St. Louis, St. Louis, MO 63110 2Pennsylavania College of Optometry, Salus University, Elkins Park, PA 19027 DOI: 10.1523/JNEUROSCI.2985-17.2018 Received: 17 October 2017 Revised: 19 January 2018 Accepted: 24 January 2018 Published: 12 February 2018 Author contributions: S.S., I.V.P., E.V.O., and A.M.D. performed research; S.S., I.V.P., V.J.K., and A.M.D. analyzed data; V.J.K. and A.M.D. designed research; V.J.K. and A.M.D. wrote the paper. Conflict of Interest: The authors declare no competing financial interests. This work was supported by NIH grants EY11522 (AMD), EY19312, EY25696, and EY27387 (VJK), EY02687 (Washington University, Department of Ophthalmology and Visual Sciences), Pennsylvania Department of Health Formula Grant (AMD) and by Research to Prevent Blindness. Correspondence should be addressed to Co-corresponding authors: Alexander M. Dizhoor, Pennsylvania College of Optometry, Salus University, Elkins Park, PA 19027, [email protected]; Vladimir J. Kefalov, Department of Ophthalmology and Visual Sciences, Washington University in St. Louis, St. Louis, MO 63110, [email protected] Cite as: J. Neurosci ; 10.1523/JNEUROSCI.2985-17.2018 Alerts: Sign up at www.jneurosci.org/cgi/alerts to receive customized email alerts when the fully formatted version of this article is published. -

IFT88 Transports Gucy2d, a Guanylyl Cyclase, to Maintain Sensory Cilia Function in Drosophila
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.15.417840; this version posted December 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Werner S et al. Title: IFT88 transports Gucy2d, a guanylyl cyclase, to maintain sensory cilia function in Drosophila Authors: Sascha Werner1,6, Sihem Zitouni1,4, Pilar Okenve-Ramos1, Susana Mendonça1,5, Anje Sporbert2, Christian Spalthoff3, Martin C. Göpfert3, Swadhin Chandra Jana1,6,7, Mónica Bettencourt-Dias1,6,7 Affiliations: 1- Instituto Gulbenkian de Ciência, Rua da Quinta Grande, nº 6, 2780-156 Oeiras, Portugal. 2- Advanced Light Microscopy, Max Delbrück Centrum for Molecular Medicine Berlin in the Helmholtz Association Robert-Rössle-Straße 10, 13125 Berlin, Germany. 3- Department of Cellular Neurobiology, University of Göttingen, 37077 Göttingen, Germany. 4- Present address: Institut de Génétique Humaine (IGH) UMR 9002 CNRS, 141 Rue de la Cardonille, Montpellier, France. 5- Present address: Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Rua Alfredo Allen, 208 4200-135 Porto, Portugal. 6- Correspondence should be addressed to Sascha Werner ([email protected]); Swadhin Chandra Jana ([email protected]); Mónica Bettencourt-Dias ([email protected]) 7- Shared lead authors. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.12.15.417840; this version posted December 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Phenotype of Autosomal Dominant Cone–Rod Dystrophy Due to the R838C Mutation of the GUCY2D Gene Encoding Retinal Guanylate
Eye (2007) 21, 1220–1225 & 2007 Nature Publishing Group All rights reserved 0950-222X/07 $30.00 www.nature.com/eye 1 2 3 3 CASE SERIES Phenotype of M Smith , N Whittock , A Searle , M Croft , C Brewer3 and M Cole1 autosomal dominant cone–rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1 Abstract should therapeutic interventions become available. Aims: To describe the phenotype of members Eye (2007) 21, 1220–1225; doi:10.1038/sj.eye.6702612; of a large Caucasian British family affected by published online 13 October 2006 autosomal dominant cone–rod dystrophy due to an R838C mutation in the guanylate cyclase Keywords: cone dystrophy; cone–rod dystrophy; 2D (GUCY2D) gene encoding retinal guanylate GUCY2D; RETGC-1 cyclase-1 (RETGC-1). Methods: Retrospective review of 29 patients from four generations of the same family. Results: Visual symptoms usually commenced Introduction in childhood. Only two patients, aged 14 and 1 Cone dystrophies are a genetically Department of 25 years, had visual acuity compatible with Ophthalmology, Torbay heterogeneous group of disorders characterised driving. Of the 12 patients aged over 40 years, Hospital, Torquay, by early deterioration of visual acuity and eight (66%) had vision of counting fingers or Devon, UK colour vision.1 Although cone dystrophies with worse and were eligible for blind registration normal rod function may occur, in most cases 2Institute of Biomedical and in the UK. Of the 29 patients, 18 (62%) had there is a degree of rod impairment, albeit less Clinical Science, Peninsula myopia greater than 5 D in at least one eye. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Exploring the Effect of Novel Small Molecules on Oligodendrocyte Precursor Proliferation Sagune Sakya University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn University Scholar Projects University Scholar Program Spring 5-1-2016 Exploring the Effect of Novel Small Molecules on Oligodendrocyte Precursor Proliferation Sagune Sakya University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/usp_projects Part of the Cellular and Molecular Physiology Commons, Medicinal Chemistry and Pharmaceutics Commons, Molecular and Cellular Neuroscience Commons, and the Nervous System Diseases Commons Recommended Citation Sakya, Sagune, "Exploring the Effect of Novel Small Molecules on Oligodendrocyte Precursor Proliferation" (2016). University Scholar Projects. 27. https://opencommons.uconn.edu/usp_projects/27 UNIVERSITY SCHOLAR PROJECT HONORS THESIS EXPLORING THE EFFECT OF NOVEL SMALL MOLECULES ON OLIGODENDROCYTE PRECURSOR PROLIFERATION By: Sagune Sakya University of Connecticut School of Pharmacy 2016 Chair and Major Project Advisor: Dr. Akiko Nishiyama, Department of Physiology and Neurobiology Advisor: Dr. Dennis Wright, Department of Pharmaceutical Sciences Advisor: Dr. Daniel Schwartz, Department of Physiology and Neurobiology Honors Advisor: Dr. Brian Aneskievich, Department of Pharmaceutical Sciences Sakya 2 Introduction Gliomas are the most common primary brain tumor in adults, representing about 81% of malignant brain tumors (Ostrom et al., 2014). Gliomas are malignant and invasive tumors that arise from glial tissue. Although it is a relatively rare disease, it can be devastating. Severe forms of glioma spread throughout the brain, destroy normal brain tissue, and are resistant to treatment. Gliomas can be classified in different ways. Gliomas can be categorized based on the predominant cell type in the tumors as astrocytomas, oligodendrogliomas, or oligoastrocytomas if the tumor has characteristics of both astrocytes and oligodendrocytes. -

1205 Domain Analysis of Human Transmembrane Guanylyl Cyclase Receptors
[Frontiers in Bioscience 10, 1205-1220 May 1, 2005] DOMAIN ANALYSIS OF HUMAN TRANSMEMBRANE GUANYLYL CYCLASE RECEPTORS: IMPLICATIONS FOR REGULATION Lincoln R. Potter Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA TABLE OF CONTENTS 1. Abstract 2. Introduction 3. Ligands, tissue expression and "knockouts" 4. Common features of transmembrane guanylyl cyclases 4.1. General topology and oligomeric state 4.2. Homology of full-length receptors 4.3. Extracellular domain 4.3.1. Glycosylation 4.3.2. Extracellular disulfide bonds 4.4. Transmembrane domain 4.5. Kinase homology domain 4.6. Dimerization domain 4.7. Catalytic domain 5. Regulation 5.1. Effect of ATP 5.2. Phosphorylation and homologous desensitization 5.3. Regulation by protein kinase c and calcium 6. Perspective 7. Acknowledgements 8. References 1. ABSTRACT In the human genome, sequence analysis review, the tissue expression, ligands and "knockout" indicates there are five functional transmembrane guanylyl phenotypes of each receptor are summarized and individual cyclases, enzymes that synthesize the intracellular second domains are compared. In the second part, regulation by messenger, cGMP. Two, GC-A and GC-B or NPR-A and ATP, calcium, protein kinase C and phosphorylation is NPR-B, are widely distributed receptors for atrial discussed. natriuretic peptide, brain natriuretic peptide and C-type natriuretic peptide, more commonly known as ANP, BNP 2. INTRODUCTION and CNP, respectively. One cyclase, GC-C or StaR, is predominantly found in the intestinal epithelium and is the Enzymes that catalyze the formation of cGMP receptor for guanylin and uroguanylin, as well as for the from GTP are called guanylyl cyclases (GCs), although bacterial pathogen, heat-stable enterotoxin (Sta). -

Novel GUCY2D Mutation Causes Phenotypic Variability of Leber Congenital Amaurosis in a Large Kindred Libe Gradstein1, Jenny Zolotushko2, Yuri V
Gradstein et al. BMC Medical Genetics (2016) 17:52 DOI 10.1186/s12881-016-0314-2 RESEARCH ARTICLE Open Access Novel GUCY2D mutation causes phenotypic variability of Leber congenital amaurosis in a large kindred Libe Gradstein1, Jenny Zolotushko2, Yuri V. Sergeev3, Itay Lavy1, Ginat Narkis2, Yonatan Perez2, Sarah Guigui1, Dror Sharon4, Eyal Banin4, Eyal Walter1, Tova Lifshitz1 and Ohad S. Birk2,5* Abstract Background: Leber congenital amaurosis (LCA) is a severe retinal degenerative disease that manifests as blindness or poor vision in infancy. The purpose of this study was to clinically characterize and identify the cause of disease in a large inbred Bedouin Israeli tribe with LCA. Methods: Thirty individuals of a single kindred, including eight affected with LCA, were recruited for this study. Patients’ clinical data and electroretinography (ERG) findings were collected. Molecular analysis included homozygosity mapping with polymorphic markers and Sanger sequencing of candidate genes. Results: Of the eight affected individuals of the kindred, nystagmus was documented in five subjects and keratoconus in three. Cataract was found in 5 of 16 eyes. Photopic and scotopic ERG performed in 5 patients were extinguished. All affected subjects were nearly blind, their visual acuity ranged between finger counting and uncertain light perception. Assuming autosomal recessive heredity of a founder mutation, studies using polymorphic markers excluded homozygosity of affected individuals at the genomic loci of all previously known genes associated with LCA, except GUCY2D.SequencingofGUCY2D identified a novel missense mutation (c.2129C>T; p.Ala710Val) resulting in substitution of alanine by valine at position 710 within the protein kinase domain of the retina-specific enzyme guanylate cyclase 1 (GC1) encoded by GUCY2D. -
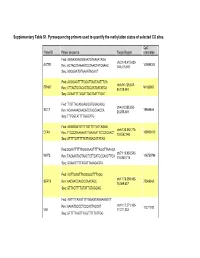
Supplementary Table S1. Pyrosequencing Primers Used to Quantify the Methylation Status of Selected CG Sites
Supplementary Table S1. Pyrosequencing primers used to quantify the methylation status of selected CG sites. CpG Probe ID Primer sequence Target Region coordinate Fwd: AGGAGGAGGGAATGTAAAATAGA chr3:148,415,620- AGTR1 149898314 Rev: ACTACCTAAAATCCTAACTATCAAAC 148,415,697 Seq: AGGGAATGTAAAATAGAGT Fwd: AGGGAATTTTGGATTAGTAATTTGA chr6:94,129,308- EPHA7 94185987 Rev: CTTACTCCACACTCCAATAATATCA 94,129,484 Seq: GGAATTTTGGATTAGTAATTTGAT Fwd: TTGTTAGAGGAGGGTGGAGAGG chr4:20,255,253- SLIT2 19864444 Rev: ACAAAAACAACATCTACCAACCA 20,255,449 Seq: TTTGGTATTTTGGGTTG Fwd: AGGGGATGTTTTGTTTTTATTAGAG chr6:133,562,774- EYA4 133603412 Rev: TTCCCRAAAAATTTAAAAATTCTCTCAACT 133,562,943 Seq: GTTTTGTTTTTATTAGAGGTATAG Fwd:GGAATTTTTAGGGAAGTTTTAGGTTAAAGA chr7:116,963,543- WNT2 116750796 Rev: TACAAATACTAACTCTTCATCCCAACTTCA 116,963,716 Seq: GGAAGTTTTAGGTTAAAGAGTA Fwd: AGTTGAGTTAGGGGGTTTAGG chr17:75,369,482- SEPT9 Rev: AACAACCAACCCAACACC 75369562 75,369,637 Seq: GTTAGTTTTGTATTGTAGGAG Fwd: AGTTTTAGGTTTYGGAGTAGGAAGGTT chr10:17,271,103- Rev: AAAATCCCCTCCCACTACCAT 17271192 VIM 17,271,233 Seq: GTTTTTAGTTYGGTTTTTATTGG Supplementary Table S2. Selected candidates: probes selected as candidates for colorectal cancer diagnostic markers based on microarray data. Normal Tumor tissue tissue Probe Name Gene cg CpG t-test Chromosome Annotation Product Mean Mean Symbol number coordinate p-value (Standard (Standard deviation) deviation) Homo sapiens angiotensin angiotensin II II receptor, type 1 receptor, AGTR1_P41_F AGTR1 cg22498996 3 149898314 0.063 (0.003) 0.481 (0.033) 2.48E-21 (AGTR1), transcript type 1 variant 1, mRNA. Homo sapiens EPH ephrin receptor EPHA7_E6_F EPHA7 cg20253378 6 94185987 receptor A7 (EPHA7), EphA7 0.057 (0.002) 0.303 (0.028) 1.27E-13 mRNA. Homo sapiens wingless- wingless-type MMTV type MMTV integration integration site family WNT2_P217_F WNT2 cg02655774 7 116750796 0.070 (0.003) 0.495 (0.037) 1.40E-19 site family member 2 member 2 precursor (WNT2), mRNA. -

GUCY2D Gene Loss-Of-Function Mutations Responsible for Leber Congenital Amaurosis 1
GUCY2D Gene Loss-of-Function Mutations Responsible for Leber Congenital Amaurosis 1 Feng Xue School of Medicine, Zhejiang University Tianying Wei Zhejiang University School of Medicine Junhui Sun Zhejiang University School of Medicine Yuqin Luo Zhejiang University School of Medicine Yanan Huo Zhejiang University School of Medicine Ping Yu Zhejiang University School of Medicine Jiao Chen Zhejiang University School of Medicine Xiaoming Wei BGT-Wuhan Ming Qi Zhejiang University School of Medicine Yinghui Ye ( [email protected] ) Women's Hospital, Zhejiang University School of Medicine https://orcid.org/0000-0001-7520-3871 Research article Keywords: Leber congenital amaurosis 1, GUCY2D, catalytic activity, 3',5'-cyclic guanosine monophosphate, guanylate cyclase 1 Posted Date: October 7th, 2019 DOI: https://doi.org/10.21203/rs.2.11649/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/17 Abstract Background: Leber congenital amaurosis (LCA) is a group of severe congenital neurodegenerative diseases. Variants in the guanylate cyclase 2D gene ( GUCY2D ), which encodes guanylate cyclase 1 (ROS-GC1), are associated with LCA1 and account for 6%–21% of all LCA cases. Methods: In this study, one family with LCA1 was recruited from China. A combination of next generation sequencing and Sanger sequencing was used to screen for disease-causing mutations. Additionally, immunohistochemistry and HPLC-coupled tandem mass spectrometry (HPLC-MS/MS) were used to conrm the cellular location and catalytic activity of ROS-GC1 mutants, respectively. Results: We found three novel mutations (c.139_139delC, c.835G>A and c.2783G>A) in the GUCY2D gene. -

Autosomal Dominant Cone-Rod Dystrophy with Mutations in the Guanylate Cyclase 2D Gene Encoding Retinal Guanylate Cyclase-1
OPHTHALMIC MOLECULAR GENETICS Autosomal Dominant Cone-Rod Dystrophy With Mutations in the Guanylate Cyclase 2D Gene Encoding Retinal Guanylate Cyclase-1 Susan M. Downes, MD; Annette M. Payne, PhD; Rosemary E. Kelsell, PhD; Frederick W. Fitzke, PhD; Graham E. Holder, PhD; David M. Hunt, PhD; Anthony T. Moore, FRCOphth; Alan C. Bird, MD Objective: To describe the phenotype in 4 families with cal testing revealed a marked loss of cone function with dominantly inherited cone-rod dystrophy, 1 with an only minimal rod involvement, even in older subjects. R838C mutation and 1 with an R838H mutation in the Photopic and scotopic static perimetry demonstrated cen- guanylate cyclase 2D (GUCY2D) gene encoding retinal tral and peripheral cone-mediated threshold elevations guanylate cyclase-1. with midperipheral sparing. Methods: Psychophysical and electrophysiological evalu- Conclusion: The phenotype associated with autosomal ation and confocal laser scanning ophthalmoscopic im- dominant cone-rod dystrophy with either an R838C or aging was performed on 10 affected members of 4 Brit- R838H mutation in GUCY2D is distinctive, with predomi- ish families. nantly cone system involvement. There is some variation in severity within the 3 families with the R838C mutation. Results: Although subjects had lifelong poor vision in bright light, a major reduction in visual acuity did not Clinical Relevance: Families with the R838C or R838H occur in most of them until after their late teens. Fun- mutation have a much milder phenotype than the fam- dus abnormalities were confined to the central macula, ily previously described that had 2 sequence changes, and increasing central atrophy was noted with age. -

Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’S Disease
SAGE-Hindawi Access to Research International Journal of Alzheimer’s Disease Volume 2010, Article ID 604792, 56 pages doi:10.4061/2010/604792 Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease Wayne Chadwick, 1 Randall Brenneman,1, 2 Bronwen Martin,3 and Stuart Maudsley1 1 Receptor Pharmacology Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA 2 Miller School of Medicine, University of Miami, Miami, FL 33124, USA 3 Metabolism Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA Correspondence should be addressed to Stuart Maudsley, [email protected] Received 17 May 2010; Accepted 27 July 2010 Academic Editor: Gemma Casadesus Copyright © 2010 Wayne Chadwick et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Various animal models of Alzheimer’s disease (AD) have been created to assist our appreciation of AD pathophysiology, as well as aid development of novel therapeutic strategies. Despite the discovery of mutated proteins that predict the development of AD, there are likely to be many other proteins also involved in this disorder. Complex physiological processes are mediated by coherent interactions of clusters of functionally related proteins. Synaptic dysfunction is one of the hallmarks of AD. Synaptic proteins are organized into multiprotein complexes in high-density membrane structures, known as lipid rafts. These microdomains enable coherent clustering of synergistic signaling proteins.