Delineating FOXG1 Syndrome from Congenital Microcephaly to Hyperkinetic Encephalopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Simple Differentiation Protocol for Generation of Induced
1 Article, Special Issue "Induced Pluripotent Stem Cells in Neurodegenerative Diseases: Application for Therapy and 2 Disease Modeling" 3 A simple differentiation protocol for generation of 4 induced pluripotent stem cell-derived basal forebrain 5 cholinergic neurons for Alzheimer’s disease and 6 frontotemporal dementia disease modeling 7 Supplemental information 8 Method 1. Reprogramming and characterisation of MBE2960 healthy control iPSC line 9 The iPSCs were generated using skin fibroblasts obtained from subjects over the age of 18 years 10 by episomal method as described [40]. Briefly, reprogramming was performed on passage 8-10 11 fibroblasts by nucleofection (Lonza Amaxa Nucleofector) with episomal vectors expressing 12 OCT4, SOX2, KLF4, L-MYC, LIN28 and shRNA against p53 [41] in feeder- and serum- free 13 conditions using TeSR-E7 medium (Stemcell Technologies). Subsequently, reprogrammed 14 colonies were manually dissected to establish clonal cell lines [42]. Three clones were assessed 15 for pluripotency markers via immunocytochemistry (Figure S1A). The iPSC line was expanded 16 and characterised. Embryoid bodies were obtained as described [43] and using tri-lineage 17 differentiation kit (Stemcell Technologies). Germ layer differentiation was assessed by 18 immunochemistry (Figure S1B). Copy number variation (CNV) analysis of original fibroblasts 19 and iPSCs from MBE2960 (Figure S1C) was performed using Illumina HumanCore Beadchip 20 arrays as we described [40]. CNV analyses were performed using PennCNV and QuantiSNP with 21 default parameter settings [44,45]. Chromosomal aberrations were deemed to involve at least 10 22 contiguous single nucleotide polymorphisms (SNPs) or a genomic region spanning at least 1MB 23 [44,45]. The B allele frequency (BAF) and the log R ratio (LRR) were extracted from 24 GenomeStudio (Illumina) for representation (Figure S1D). -

Diagnosis of FOXG1 Syndrome Caused by Recurrent Balanced Chromosomal Rearrangements: Case Study and Literature Review Connor P
Craig et al. Mol Cytogenet (2020) 13:40 https://doi.org/10.1186/s13039-020-00506-1 CASE REPORT Open Access Diagnosis of FOXG1 syndrome caused by recurrent balanced chromosomal rearrangements: case study and literature review Connor P. Craig1,2, Emily Calamaro3, Chin‑To Fong3, Anwar M. Iqbal1, Alexander R. Paciorkowski3,4,5,6 and Bin Zhang1,3* Abstract Background: The FOXG1 gene plays a vital role in mammalian brain diferentiation and development. Intra‑ and intergenic mutations resulting in loss of function or altered expression of the FOXG1 gene cause FOXG1 syndrome. The hallmarks of this syndrome are severe developmental delay with absent verbal language, post‑natal growth restric‑ tion, post‑natal microcephaly, and a recognizable movement disorder characterized by chorea and dystonia. Case presentation: Here we describe a case of a 7‑year‑old male patient found to have a de novo balanced translo‑ cation between chromosome 3 at band 3q14.1 and chromosome 14 at band 14q12 via G‑banding chromosome and Fluorescence In Situ Hybridization (FISH) analyses. This rearrangement disrupts the proximity of FOXG1 to a previously described smallest region of deletion overlap (SRO), likely resulting in haploinsufciency. Conclusions: This case adds to the growing body of literature implicating chromosomal structural variants in the manifestation of this disorder and highlights the vital role of cis‑acting regulatory elements in the normal expression of this gene. Finally, we propose a protocol for refex FISH analysis to improve diagnostic efciency for patients with suspected FOXG1 syndrome. Keywords: FOXG1, Haploinsufciency, Postnatal microcephaly, FISH, Enhancer, Chromosomal rearrangement, Diagnosis Introduction brain development, with high levels of expression in the Te Forkhead Box G1 (FOXG1) gene [OMIM: 164874], developing fetal telencephalon [1–4]. -

Notch Signal Controls Several Steps of Inner Ear Development Norio Yamamoto and Matthew W
NIDCD Notch signal controls several steps of inner ear development Norio Yamamoto and Matthew W. Kelley Section on Developmental Neuroscience Section on Developmental Neuroscience National Institute on Deafness and Other Communication Disorders National Institutes of Health Abstract Problem addressed RBP-J mutant cochleae did not have any supporting cells Notch signaling has been reported to contribute to inner ear development, however, its specific functions remain unclear, partly because of discrepancies between the phenotypes of mutant mice with single deletion of specific Notch related genes. These discrepancies are probably due to functional compensation by other Notch Figure 4 receptors or ligands. Foxg1 Cre;RBP-J floxed/+ Foxg1 Cre;RBP-J floxed/floxed A B C D To determine the effects of complete elimination of Notch signaling, we used a conditional knockout of the Rbpsuh gene. RBP-J protein is a critical transcriptional p27 Phalloidin p27 Phalloidin To test if mutant cochleae contained supporting cells we co-activator for all Notch molecules and thus deletion of this protein inhibits all Notch signaling. examined expression of supporting cell markers such as p27 and Prox1. On E17.5 p27 was expressed in supporting cells Methods and Measures ***** ***** under or between hair cells such as inner pharyngeal cells, Floxed Rbpsuh mice were crossed with Foxg1-Cre knock-in mice to delete the Rbpsuh gene in the inner ear. Inner ear phenotypes in Rbpsuh conditional knockout Deiter's cells and pillar cells (asterisks in figure 4 A and B). mice were determined at various developmental stages using immunohistochemistry. But no p27 expression was detected in those regions of RBP-J E Prox1 F Phalloidin G Prox1 H Phalloidin conditional knockout mice (Figure 4 C and D). -

Downloaded from Bioscientifica.Com at 09/26/2021 08:38:14PM Via Free Access
1 185 L C Gregory, P Gergics, OTX2 mutations in congenital 185:1 121–135 Clinical Study M Nakaguma and others hypopituitarism The phenotypic spectrum associated with OTX2 mutations in humans Louise C Gregory 1,*, Peter Gergics2,* , Marilena Nakaguma3,* , Hironori Bando2 , Giuseppa Patti1,4,5, Mark J McCabe1, Qing Fang 2, Qianyi Ma2 , Ayse Bilge Ozel2 , Jun Z Li2 , Michele Moreira Poina3, Alexander A L Jorge 3, Anna F Figueredo Benedetti3, Antonio M Lerario3, Ivo J P Arnhold3 , Berenice B Mendonca3 , Mohamad Maghnie4,5, Sally A Camper2 , Luciani R S Carvalho3 and Mehul T Dattani1 1Section of Molecular Basis of Rare Disease, Genetics and Genomic Medicine Research & Teaching Department, UCL Great Ormond Street Institute of Child Health, London, UK, 2Department of Human Genetics, University of Michigan, Correspondence Ann Arbor, Michigan, USA, 3Developmental Endocrinology Unit, Hospital das Clinicas da Faculdade de Medicina da should be addressed Universidade de São Paulo, São Paulo, Brazil, 4Department of Pediatrics, IRCCS Istituto Giannina Gaslini and to S A Camper or M T 5Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Dattani Genova, Genova, Italy Email *(L C Gregory, P Gergics and M Nakaguma contributed equally to this work) [email protected] or [email protected] Abstract Objective: The transcription factor OTX2 is implicated in ocular, craniofacial, and pituitary development. Design: We aimed to establish the contribution of OTX2 mutations in congenital hypopituitarism patients with/without eye abnormalities, study functional consequences, and establish OTX2 expression in the human brain, with a view to investigate the mechanism of action. Methods: We screened patients from the UK (n = 103), international centres (n = 24), and Brazil (n = 282); 145 were within the septo-optic dysplasia spectrum, and 264 had no eye phenotype. -
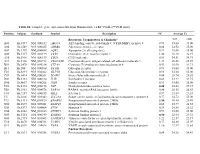
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

14Q12q13.3 Deletion Diagnosed Using Chromo Somal Microarray
Case Report Neonatal Med 2020 November;27(4):207-213 https://doi.org/10.5385/nm.2020.27.4.207 pISSN 2287-9412 . eISSN 2287-9803 14q12q13.3 Deletion Diagnosed Using Chromo somal Microarray Analysis in an Infant Showing Seizures, Hypoplasia of the Corpus Callosum, and Developmental Delay Jae Hyuk Kwon, MD1, Young Hwa Song, MD1, Jung Min Yoon, MD1, Eun Jung Cheon, MD1, Kyung Ok Ko, MD1, Jae Woo Lim, MD1, and Hyon J. Kim, MD, FACMG2 Departments of 1Pediatrics and 2Medical Genetics, Konyang University College of Medicine, Daejeon, Korea ABSTRACT Received: 18 August 2020 Revised: 25 September 2020 14q12q13.3 Deletion is a rare microdeletion syndrome associated with neurodevelop Accepted: 6 October 2020 mental delay, failure to thrive, seizures, and abnormal brain development. Symptoms Correspondence to: Hyon J. Kim, MD vary depending on the sites of gene deletion, and establishing the diagnosis is often Department of Medical Genetics, difficult, as the condition cannot be detected with routine chromosome analysis. In Konyang University College of Medi this report, we present a patient with intrauterine growth retardation, microcephaly, cine, 158 Gwanjeodongro, Seogu, muscle weakness, seizures, and hypoplasia of the corpus callosum who underwent Daejeon 35365, Korea diagnostic tests, including karyotyping in the neonatal period without leading to a Tel: +82426009230 specific diagnosis. The patient was confirmed with a serious developmental disorder, Fax: +82426009090 and a chromosomal microarray analysis was performed at 8 months of age, revealing Email: [email protected] a 14q12q13.3 deletion. In this case, the condition was diagnosed in early infancy, in contrast to previously reported cases, and the patient had diverse and severe symptoms. -

Grimme, Acadia.Pdf
MECHANISM OF ACTION OF HISTONE DEACETYLASE INHIBITORS ON SURVIVAL MOTOR NEURON 2 PROMOTER by Acadia L. Grimme A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Bachelors of Science in Biological Sciences with Distinction Spring 2018 © 2018 Acadia Grimme All Rights Reserved MECHANISM OF ACTION OF HISTONE DEACETYLASE INHIBITORS ON SURVIVAL MOTOR NEURON 2 PROMOTER by Acadia L. Grimme Approved: __________________________________________________________ Matthew E. R. Butchbach, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Deni S. Galileo, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Carlton R. Cooper, Ph.D. Committee member from the Department of Biological Sciences Approved: __________________________________________________________ Gary H. Laverty, Ph.D. Committee member from the Board of Senior Thesis Readers Approved: __________________________________________________________ Michael Chajes, Ph.D. Chair of the University Committee on Student and Faculty Honors ACKNOWLEDGMENTS I would like to acknowledge my thesis director Dr. Butchbach for his wonderful guidance and patience as I worked through my project. He has been an excellent research mentor over the last two years and I am forever thankful that he welcomed me into his lab. His dedication to his work inspires me as an aspiring research scientist. His lessons will carry on with me as I pursue future research in graduate school and beyond. I would like to thank both current and former members of the Motor Neuron Disease Laboratory: Sambee Kanda, Kyle Hinkle, and Andrew Connell. Sambee and Andrew patiently taught me many of the techniques I utilized in my project, and without them it would not be what it is today. -

Deficits in Human Trisomy 21 Ipscs and Neurons
Deficits in human trisomy 21 iPSCs and neurons Jason P. Weicka,1, Dustie L. Helda, George F. Bonadurer IIIa, Matthew E. Doersa, Yan Liua, Chelsie Maguirea, Aaron Clarka, Joshua A. Knackerta, Katharine Molinaroloa, Michael Mussera, Lin Yaoa, Yingnan Yina, Jianfeng Lua, Xiaoqing Zhanga, Su-Chun Zhanga,b,c,d, and Anita Bhattacharyyaa,2 aWaisman Center, bNeuroscience Training Program, and Departments of cNeuroscience and dNeurology, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53705 Edited by Fred H. Gage, The Salk Institute for Biological Studies, San Diego, CA, and approved May 1, 2013 (received for review September 26, 2012) Down syndrome (trisomy 21) is the most common genetic cause of uniparental disomy (or isodisomy), which is often associated with intellectual disability, but the precise molecular mechanisms un- trisomy rescue (Fig. 1D). Short tandem repeats at various loci derlying impaired cognition remain unclear. Elucidation of these indicated that the Ts21 and euploid lines were isogenic other than mechanisms has been hindered by the lack of a model system that the presence of Ts21 (Fig. 1E). To increase statistical power, contains full trisomy of chromosome 21 (Ts21) in a human genome fibroblasts from a second DS individual were reprogrammed us- that enables normal gene regulation. To overcome this limitation, we ing Sendai virus (12), an RNA virus, which yielded another Ts21 created Ts21-induced pluripotent stem cells (iPSCs) from two sets of iPSC line (Fig. 1C). All iPSC clones were named according to Ts21 human fibroblasts. One of the fibroblast lines had low level standardized naming procedures (Fig. 1F) (13) and used in all mosaicism for Ts21 and yielded Ts21 iPSCs and an isogenic control experiments. -

Improving Tissue Differentiation in Cerebral Organoids
Improving tissue differentiation in cerebral organoids Technical Journal Club Manfredi Carta 16.06.2020 Contents Introduction Protocol overview 1. hPSCs are incubated in human ES medium with low bFGF4 and ROCK inhibitor embryoid bodies (= 3D aggregates of hPSCs) 2. Transfer to neural induction medium (DMEM/F12, N2, GluMax, amino acids, heparin) 3. Transfer to Matrigel, grow in differentiation media (DMEM/F12, Neurobasal, B27 w/o Vitamin A, N2, β-ME, insulin, GluMax, amino acids) 4. Add Vitamin A to media, grow in spinning bioreactor bFGF4: basic fibrobl. growth factor 4 ROCK: Rho-associated protein kinase Spinning conditions Tissues grown in spinning bioreactor • developed contained large fluid-filled cavities (similar to ventricles) • apical localisation of neural N-cadherin • Were larger and more continuous than tissues grown in stationary suspension Fluid-filled cavity Retina-like pigmented epithelium SOX2+: neural progenitors Scale bars: 200µm TUJ1+: neurons Cerebral organoids: Anatomy Forebrain Choroid plexus Molecular layer Meninges Choroid plexus • Spinning conditions: anatomy reminiscent of brain regions • Organoids lacked vasculature, limited oxygen and nutrient diffusion: cell death in internal areas (TUNEL stain) Mammalian brain development • Neuroepithelium expands to generate radial glial stem cells (RG) • RG divide at the apical surface within the ventricular zone (VZ), generate neurons and intermediate progenitor cells (IPCs) • IPCs populate the subventricular zone (SVZ), neurons migrate through intermediate zone (IZ) to -

Xo PANEL DNA GENE LIST
xO PANEL DNA GENE LIST ~1700 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes (at 400 -500x average coverage on tumor) Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 -

Sensory Cortex Wiring Requires Preselection of Short- and Long-Range Projection Neurons Through an Egr-Foxg1-COUP-TFI Network
ARTICLE https://doi.org/10.1038/s41467-019-11043-w OPEN Sensory cortex wiring requires preselection of short- and long-range projection neurons through an Egr-Foxg1-COUP-TFI network Pei-Shan Hou1,2,3, Goichi Miyoshi 4 & Carina Hanashima1,2,3 The bimodal requisite for a genetic program and external stimuli is a key feature of sensory circuit formation. However, the contribution of cell-intrinsic codes to directing sensory- 1234567890():,; specific circuits remains unknown. Here, we identify the earliest molecular program that preselects projection neuron types in the sensory neocortex. Mechanistically, Foxg1 binds to an H3K4me1-enriched enhancer site to repress COUP-TFI, where ectopic acquisition of Foxg1 in layer 4 cells transforms local projection neurons to callosal projection neurons with pyr- amidal morphologies. Removal of Foxg1 in long-range projection neurons, in turn, derepresses COUP-TFI and activates a layer 4 neuron-specific program. The earliest segregation of projection subtypes is achieved through repression of Foxg1 in layer 4 precursors by early growth response genes, the major targets of the transforming growth factor-β signaling pathway. These findings describe the earliest cortex-intrinsic program that restricts neuronal connectivity in sensory circuits, a fundamental step towards the acquisition of mammalian perceptual behavior. 1 Department of Biology, Faculty of Education and Integrated Arts and Sciences, Waseda University, Shinjuku-ku, Tokyo 162-8480, Japan. 2 Graduate School of Advanced Science and Engineering, Waseda University, Shinjuku-ku, Tokyo 162-8480, Japan. 3 Laboratory for Neocortical Development, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan. 4 Department of Neurophysiology, Tokyo Women’s Medical University School of Medicine, Shinjuku-ku, Tokyo 162-8666, Japan. -

The Dynamic Interplay Between Homeodomain Transcription Factors and Chromatin Environment Regulates Proneural Factor Outcomes
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.02.398677; this version posted December 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Ang et al pg.1 THE DYNAMIC INTERPLAY BETWEEN HOMEODOMAIN TRANSCRIPTION FACTORS AND CHROMATIN ENVIRONMENT REGULATES PRONEURAL FACTOR OUTCOMES Cheen Euong Ang1,2,3, Victor Hipolito Olmos2,3, Bo Zhou2,3,4, Qian Yi Lee1,2,3, Rahul Sinha3, Aadit Narayanaswamy2,3, Moritz Mall2,3, Kirill Chesnov2,3, Thomas Sudhof4 and Marius Wernig2,3 1. Department of Bioengineering 2. Department of Pathology 3. Institute of Stem Cell and Regenerative Medicine 4. Department of Molecular and Cellular Physiology and Howard Hughes Medical Institute Stanford University, Stanford, CA 94305, USA Correspondence to: [email protected] (M.W.) bioRxiv preprint doi: https://doi.org/10.1101/2020.12.02.398677; this version posted December 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Ang et al pg.2 SUMMARY Generation of neurons of vast diversity involves early spatial and temporal patterning of the neuronal precursors by morphogenic gradients and combinatorial expression of transcription factors. While the proneuronal function of the basic-helix-loop-helix (bHLH) transcription factor Ngn2 is well established, its role in neuronal subtype specification remains unclear. Here, we found that coexpressing NGN2 with the forebrain homeobox factor EMX1 converts human pluripotent stem cells into a highly homogeneous glutamatergic forebrain neurons without partial cholinergic and monoaminergic gene programs observed in cells infected with NGN2 only.