A Human Multi-Lineage Hepatic Organoid Model for Liver Fibrosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FLT3 Inhibitors in Acute Myeloid Leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2*
Wu et al. Journal of Hematology & Oncology (2018) 11:133 https://doi.org/10.1186/s13045-018-0675-4 REVIEW Open Access FLT3 inhibitors in acute myeloid leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2* Abstract FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents. Keywords: FMS-like tyrosine kinase 3 inhibitors, Acute myeloid leukemia, Midostaurin, FLT3 Introduction RAS, MEK, and PI3K/AKT pathways [10], and ultim- Acute myeloid leukemia (AML) remains a highly resist- ately causes suppression of apoptosis and differentiation ant disease to conventional chemotherapy, with a me- of leukemic cells, including dysregulation of leukemic dian survival of only 4 months for relapsed and/or cell proliferation [11]. refractory disease [1]. Molecular profiling by PCR and Multiple FLT3 inhibitors are in clinical trials for treat- next-generation sequencing has revealed a variety of re- ing patients with FLT3/ITD-mutated AML. In this re- current gene mutations [2–4]. New agents are rapidly view, we summarized the preclinical and clinical studies emerging as targeted therapy for high-risk AML [5, 6]. on new FLT3 inhibitors, including sorafenib, lestaurtinib, In 1996, FMS-like tyrosine kinase 3/internal tandem du- sunitinib, tandutinib, quizartinib, midostaurin, gilteriti- plication (FLT3/ITD) was first recognized as a frequently nib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG mutated gene in AML [7]. -

Precision Medicine in Pediatric Neurooncology: a Review
UCLA UCLA Previously Published Works Title Precision Medicine in Pediatric Neurooncology: A Review. Permalink https://escholarship.org/uc/item/6tv2b43d Journal ACS chemical neuroscience, 9(1) ISSN 1948-7193 Authors Mochizuki, Aaron Y Frost, Isaura M Mastrodimos, Melina B et al. Publication Date 2018 DOI 10.1021/acschemneuro.7b00388 Peer reviewed eScholarship.org Powered by the California Digital Library University of California HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author ACS Chem Manuscript Author Neurosci. Author Manuscript Author manuscript; available in PMC 2019 July 24. Published in final edited form as: ACS Chem Neurosci. 2018 January 17; 9(1): 11–28. doi:10.1021/acschemneuro.7b00388. Precision Medicine in Pediatric Neurooncology: A Review Aaron Y. Mochizuki∥, Isaura M. Frost∥, Melina B. Mastrodimos∥, Ashley S. Plant┴, Anthony C. Wang#, Theodore B. Moore∥, Robert M. Prins#,∇,○, Paul S. Weiss*,†,‡,§,∇, Steven J. Jonas*,†,∥,◆,¶ †California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, United States ‡Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California 90095, United States §Department of Materials Science and Engineering, University of California, Los Angeles, Los Angeles, California 90095, United States ∥Department of Pediatrics, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California 90095, United States ┴Division of Pediatric Oncology, Children’s Hospital of Orange -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Crenolanib Is a Selective Type I Pan-FLT3 Inhibitor
Crenolanib is a selective type I pan-FLT3 inhibitor Catherine Choy Smitha,b,1, Elisabeth A. Lasatera,1, Kimberly C. Lina, Qi Wangc, Melissa Quino McCreeryd, Whitney K. Stewarta, Lauren E. Damona, Alexander E. Perle, Grace R. Jeschkee, Mayumi Sugitae, Martin Carrolle, Scott C. Koganb,d, John Kuriyanc, and Neil P. Shaha,b,2 aDivision of Hematology/Oncology, bHelen Diller Family Comprehensive Cancer Center, and dDivision of Laboratory Medicine, University of California, San Francisco, CA 94143; cDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; and eDivision of Hematology/Oncology, Perelman School of Medicine and the Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA 19104 Edited* by Charles L. Sawyers, Memorial Sloan–Kettering Cancer Center, New York, NY, and approved February 13, 2014 (received for review November 4, 2013) Tyrosine kinase inhibitors (TKIs) represent transformative thera- inhibitor and holds significant promise for achieving prolonged pies for several malignancies. Two critical features necessary for remissions. However, ponatinib potently inhibits a number of maximizing TKI tolerability and response duration are kinase selec- kinases, which may be responsible for unanticipated serious tivity and invulnerability to resistance-conferring kinase domain (KD) cardiovascular toxicities that have emerged with this agent. mutations in the intended target. No prior TKI has demonstrated The ability to selectively suppress resistance-conferring muta- both of these properties. -

Pdgfra) D842v Mutant Gastrointestinal Stromal Tumor (Gist
PRELIMINARY REPORT OF CRENOLANIB IN THE TREATMENT OF ADVANCED PLATELET DERIVED GROWTH FACTOR A (PDGFRA) D842V MUTANT GASTROINTESTINAL STROMAL TUMOR (GIST) M. von Mehren1, M. Heinrich2, E. Tetzlaff1, K. Padavic-Shaller1, M. Yu1, C. Muralidhara3, A.Ramachandran3, H. Shah3 1Fox Chase Cancer Center, Philadelphia, PA; 2Oregon Health and Science University, Portland, OR; 3AROG Pharmaceuticals, Dallas, TX. BACKGROUND PATIENT CHARACTERISTICS PHARMACOKINETICS PARTIAL METABOLIC RESPONSE Many patients with advanced GIST treated with • To date, 7 patients (4 F, 3 M) have been accrued. • Serum pharmacokinetics samples were obtained pre dose approved tyrosine kinase therapies have prolonged • All had metastatic disease in liver and/or and at 30 (± 10), 60 (± 15), 120 (± 15) minutes and at 4 (±1), disease control with a median survival of 5 years. Rare mesentery/retroperitoneum. 8 (±2), and 24 (±4) hours after crenolanib administration subsets of GIST do not derive the same benefit from • Best response to prior therapy was stable disease. • Analysis was performed by an isocratic high performance treatment. One such subset is GIST that carries a • Safety data is available in 6 patients and efficacy in 6 patients. liquid chromatography assay with tandem mass mutation in PDGFRA exon 18, D842V. In vitro, approved spectrometry therapies do not cause a decrease in cell proliferation or • Crenolanib was rapidly absorbed, with a tmax of ~2 hours loss of PDGFRA phosphorylation1. In clinical trials, • Serum trough concentrations of crenolanib (at 24hrs) were available data suggests no response to standard ~12% the peak concentration. 2 therapies . 2000.00 1800.00 3 Crenolanib is a benzimidazole compound being 1600.00 • 2 patients had metabolically active disease (SUV ≥ developed for the treatment of GIST patients with 1400.00 D842V Pt-001 10 in baseline PET scan) upon study entry. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172915 A1 Ni (43) Pub
US 20170172915A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172915 A1 Ni (43) Pub. Date: Jun. 22, 2017 (54) COMPOSITIONS AND METHODS FOR A6IR 9/08 (2006.01) TREATING PTERYGUM A6DF 9/00 (2006.01) (71) Applicant: Cloudbreak Therapeutics, LLC, A63L/404 (2006.01) Irvine, CA (US) (52) U.S. Cl. (72) Inventor: Jinsong Ni, Irvine, CA (US) CPC .......... A61K 9/0048 (2013.01); A61F 9/0017 (2013.01); A61 K3I/496 (2013.01); A61 K (21) Appl. No.: 15/375,820 31/404 (2013.01); A61K 31/407 (2013.01); (22) Filed: Dec. 12, 2016 A61K 9/08 (2013.01); A61K 9/107 (2013.01); A61K 9/0051 (2013.01); A61 F 2210/0004 Related U.S. Application Data (2013.01) (63) Continuation of application No. PCT/US2016/ 035726, filed on Jun. 3, 2016. (57) ABSTRACT (60) Provisional application No. 62/172,063, filed on Jun. 6, 2015, provisional application No. 62/186,660, filed on Jun. 30, 2015. Compositions and methods for inducing pterygium regres sion from visual axis/central cornea, stabilizing pterygium, Publication Classification treating hyperemia and symptoms in pterygium patients, and (51) Int. Cl. treating pterygium recurrence following pterygiectomy are 3. e96 3:08: disclosed. The methods include administration of a multi A 6LX 9/07 (2006.01) kinase inhibitor, an antimetabolite or a combination thereof A6 IK 3/407 (2006.01) to patients in need thereof. Patent Application Publication Jun. 22, 2017 Sheet 1 of 5 US 2017/0172915 A1 tay is average area with Six is ws wei: *88,33;** {{.{33; ***xt.txs 3.338& 8. -
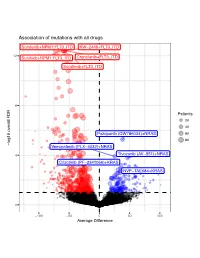
Association of Mutations with All Drugs
Association of mutations with all drugs Sorafenib+NPM1:FLT3_ITD KW−2449+FLT3_ITD 12 Sunitinib+NPM1:FLT3_ITD Crenolanib+FLT3_ITD Sorafenib+FLT3_ITD 8 Patients 20 40 Pazopanib (GW786034)+NRAS 60 80 log10 overall FDR log10 overall − Vemurafenib (PLX−4032)+NRAS 4 Tivozanib (AV−951)+NRAS Crizotinib (PF−2341066)+KRAS NVP−TAE684+KRAS 0 −100 −50 0 50 100 Average Difference Association of mutations with 17−AAG (Tanespimycin) FLT3_ITD 6 NPM1:FLT3_ITD 4 Patients 20 40 TP53 log10 FDR 60 − FLT3_ITD:DNMT3A NPM1:FLT3_ITD:DNMT3A NPM1 2 FLT3_ITD:TET2 FLT3_ITD:ASXL1 NPM1:FLT3_ITD:TET2 FLT3_ITD:WT1 TP53:NRAS FLT3_ITD:IDH2 NPM1:DNMT3A FLT3_ITD:RUNX1 0 −50 −25 0 25 50 Average Difference Association of mutations with A−674563 NPM1:FLT3_ITD FLT3_ITD 4 3 Patients 20 40 2 FLT3_ITD:TET2 60 log10 FDR − 80 NPM1:FLT3_ITD:DNMT3A NPM1 FLT3_ITD:DNMT3A FLT3_ITD:IDH2 NPM1:IDH2 CREBBP 1 NPM1:FLT3_ITD:TET2 FLT3_ITD:WT1 0 −60 −30 0 30 60 Average Difference Association of mutations with ABT−737 1.00 0.75 Patients 10 20 0.50 30 log10 FDR 40 − 50 0.25 0.00 −50 −25 0 25 Average Difference Association of mutations with AT7519 1.00 0.75 Patients 20 0.50 40 log10 FDR 60 − 0.25 0.00 −25 0 25 Average Difference Association of mutations with AZD1480 FLT3_ITD 3 NPM1:FLT3_ITD NPM1:SRSF2:IDH2 2 Patients 20 40 60 log10 FDR − NPM1:SRSF2 FLT3_ITD:TET2 80 1 FLT3_ITD:IDH2 0 −60 −40 −20 0 20 Average Difference Association of mutations with Afatinib (BIBW−2992) 1.00 0.75 Patients 20 0.50 40 60 log10 FDR − 80 0.25 0.00 −20 0 20 40 Average Difference Association of mutations with Alisertib (MLN8237) -

A Genome-Wide CRISPR Screen Identifies Genes Critical for Resistance to FLT3 Inhibitor AC220
Published OnlineFirst June 16, 2017; DOI: 10.1158/0008-5472.CAN-16-1627 Cancer Therapeutics, Targets, and Chemical Biology Research A Genome-Wide CRISPR Screen Identifies Genes Critical for Resistance to FLT3 Inhibitor AC220 Panpan Hou1,2, Chao Wu2, Yuchen Wang2,3, Rui Qi2, Dheeraj Bhavanasi4, Zhixiang Zuo5, Cedric Dos Santos6, Shuliang Chen7, Yu Chen1, Hong Zheng8, Hong Wang9, Alexander Perl4, Deyin Guo1, and Jian Huang2 Abstract Acute myeloid leukemia (AML) is a malignant hematopoietic Wnt signaling antagonist, and demonstrated reactivation of disease and the most common type of acute leukemia in adults. downstream FGF/Ras/ERK and Wnt signaling as major mechan- The mechanisms underlying drug resistance in AML are poorly isms of resistance to AC220. We confirmed these findings in understood. Activating mutations in FMS-like tyrosine kinase 3 primary AML patient samples. Expression of SPRY3 and GSK3A (FLT3) are the most common molecular abnormality in AML. was dramatically reduced in AC220-resistant AML samples, and Quizartinib (AC220) is a potent and selective second-generation SPRY3-deleted primary AML cells were resistant to AC220. inhibitor of FLT3. It is in clinical trials for the treatment of relapsed Intriguingly, expression of SPRY3 was greatly reduced in GSK3 or refractory FLT3-ITD–positive and –negative AML patients and knockout AML cells, which positioned SPRY3 downstream of as maintenance therapy. To understand the mechanisms of drug GSK3 in the resistance pathway. Taken together, our study resistance to AC220, we undertook an unbiased approach with a identified novel genes whose loss of function conferred resis- novel CRISPR-pooled library to screen new genes whose loss of tance to a selective FLT3 inhibitor, providing new insight into function confers resistance to AC220. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

Current Challenges and Opportunities in Treating Glioblastomas
Supplemental Material can be found at: /content/suppl/2018/04/23/70.3.412.DC1.html 1521-0081/70/3/412–445$35.00 https://doi.org/10.1124/pr.117.014944 PHARMACOLOGICAL REVIEWS Pharmacol Rev 70:412–445, July 2018 Copyright © 2018 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. ASSOCIATE EDITOR: ERIC L. BARKER Current Challenges and Opportunities in Treating Glioblastomas Andrea Shergalis, Armand Bankhead, III, Urarika Luesakul, Nongnuj Muangsin, and Nouri Neamati Department of Medicinal Chemistry, College of Pharmacy, North Campus Research Complex, Ann Arbor, Michigan (A.S., U.L., N.N.); Biostatistics Department and School of Public Health, University of Michigan, Ann Arbor, Michigan (A.B.); and Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, Thailand (U.L., N.M.) Abstract ...................................................................................413 I. Introduction . ..............................................................................413 II. Current Treatment Options for Glioblastoma ...............................................414 III. Molecular Diagnostic Signature of Glioblastoma . ..........................................419 IV. Characteristics of Protein Expression in Glioblastoma . .....................................419 V. Emerging Targets in Glioblastoma . ......................................................422 A. Biomarker Identification . ............................................................422 B. -

American Society of Clinical Oncology 47Th Annual Meeting
American Society of Clinical Oncology 47th Annual Meeting Meeting Program Note: Sessions in which Special Awards are presented are shaded in gray Friday, June 3, 2011 1:00 PM - 2:15 PM FRIDAY EDUCATION SESSIONS Customized Cancer Treatment: A Systems Biology Approach to Drug Selection Location: E354b CME credit: 1.25 Track(s): Developmental Therapeutics, Cancer Genetics, Clinical Trials John Mendelsohn, MD—Chair University of Texas M. D. Anderson Cancer Center Customized Cancer Medicine: Are We Ready for What It Will Take? Jeffrey M. Trent, PhD TGen Large-scale, Multidimensional Genomics Data: Making Sense for Patients Andrea Califano, PhD Columbia University Cancer’s Complexity: Are We Looking at the Wrong Levels to Develop Effective Interventions? Endometrial Cancer: Present State and Future Promise Location: E354a CME credit: 1.25 Track(s): Gynecologic Cancer Don S. Dizon, MD—Chair Warren Alpert Medical School of Brown University Options in the Treatment of Relapsed Endometrial Cancer: Beyond First-line Therapy Karen H. Lu, MD University of Texas M. D. Anderson Cancer Center Molecular Characterization of Endometrial Cancer: Going beyond Histology David Scott Miller, MD University of Texas Southwestern Medical Center Incorporation of Adjuvant Chemotherapy into Endometrial Cancer Treatment Neoadjuvant Therapy in Operable Breast Cancer: Who, What, When, Why, and How? Location: Hall D1 CME credit: 1.25 Track(s): Breast Cancer Luca Gianni, MD—Chair Fondazione IRCCS Istituto Nazionale dei Tumori Neoadjuvant Assumptions and Application to Chemotherapy, Endocrine Therapy, and Biologic Therapy-based Regimens David W. Ollila, MD University of North Carolina at Chapel Hill Surgical Implications of the Neoadjuvant Approach: Facilitating Research Biopsies, Breast Conservation in Previously T3–4 Tumors, and Axillary Staging—Before or After? Thomas A. -

Multi-Discipline Review
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 212608Orig1s000 MULTI-DISCIPLINE REVIEW Summary Review Office Director Cross Discipline Team Leader Review Clinical Review Non-Clinical Review Statistical Review Clinical Pharmacology Review NDA/BLA Multi-disciplinary Review and Evaluation NDA 212608 AYVAKIT (avapritinib) NDA/BLA Multi-Disciplinary Review and Evaluation Application Type New Drug Application (NDA)/New Molecular Entity (NME) Application Number(s) 212608 Priority or Standard Priority Submit Date(s) June 14, 2019 Received Date(s) June 14, 2019 PDUFA Goal Date February 14, 2020 Division/Office DO3/OOD Established/Proper Name Avapritinib (Proposed) Trade Name AYVAKIT Pharmacologic Class Kinase inhibitor Code name BLU-285 Applicant Blueprint Medicines Corporation Dosage form 100 mg, 200 mg, and 300 mg tablets Applicant proposed Dosing 300 mg orally once daily Regimen Applicant Proposed For the treatment of adult patients with unresectable or Indication(s)/Population(s) metastatic gastrointestinal stromal tumor (GIST) who have a platelet-derived growth factor receptor alpha (PDGFA) exon 18 mutation, regardless of prior therapy Applicant Proposed SNOMED CT Indication Gastrointestinal Stromal Tumor Disease Term for each Proposed Indication Recommendation on Approval Regulatory Action Recommended For the treatment of adults with unresectable or metastatic Indication(s)/Population(s) GIST harboring a platelet-derived growth factor receptor alpha (if applicable) (PDGFRA) exon 18 mutation, including PDGFRA D842V mutations Recommended SNOMED CT Indication Disease Gastrointestinal Stromal Tumor Term for each Indication (if applicable) Recommended Dosing 300 mg orally once daily Regimen 1 Version date: April 2, 2018 Reference ID: 4543562 NDA/BLA Multi-disciplinary Review and Evaluation NDA 212608 AYVAKIT (avapritinib) Table of Contents Table of Tables ...............................................................................................................................