Crenolanib Is a Selective Type I Pan-FLT3 Inhibitor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Human Multi-Lineage Hepatic Organoid Model for Liver Fibrosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278473; this version posted September 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A Human Multi-Lineage Hepatic Organoid Model for Liver Fibrosis Yuan Guan1, Annika Enejder2, Meiyue Wang1, Zhuoqing Fang1, Lu Cui3, Shih-Yu Chen4, Jingxiao Wang1, Yalun Tan1, Manhong Wu1, Xinyu Chen1, Patrik K. Johansson2, Issra Osman1, Koshi Kunimoto3, Pierre Russo5, Sarah C. Heilshorn2 and Gary Peltz1* 1Department of Anesthesia, Stanford University School of Medicine, Stanford CA, 94305; 2Department of Materials Science and Engineering, Stanford University, Stanford CA, 94305; 3Department of Pathology, Institute of Stem Cell Biology and Regenerative Medicine (ISCBRM), Stanford University School of Medicine, Stanford, 94305, CA, USA; 4Shih-Yu Chen, Institute of Biomedical Sciences, Academia Sinica, Taipei, 11529, Taiwan; 5Perelman School of Medicine at The University of Pennsylvania, Philadelphia, PA 19104 The authors have declared that no conflict of interest exists. *Address Correspondence to: [email protected] 300 Pasteur Dr. Room L232 Stanford, CA 94305. bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278473; this version posted September 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Despite its devastating consequences, liver fibrosis has no treatments. Genome engineering and a hepatic organoid system was used to produce the first in vitro model for human liver fibrosis. Hepatic organoids engineered to express the most common causative mutation for Autosomal Recessive Polycystic Kidney Disease (ARPKD) developed the key features of ARPKD liver pathology (abnormal bile ducts and hepatic fibrosis) in only 21 days. -

A Phase II, Open-Label Study of Ponatinib, a Multi-Targeted Oral
A Phase II, open-label study of ponatinib, a multi-targeted oral tyrosine kinase inhibitor, in advanced non- small cell lung cancer harboring RET translocations Version 8/22/2018 A Phase II, open-label study of ponatinib, a multi-targeted oral tyrosine kinase inhibitor, in advanced non-small-cell lung cancer harboring RET translocations NCT01813734 Protocol ID: 13-103 Version Date: August 22, 2018 1 Confidential A Phase II, open-label study of ponatinib, a multi-targeted oral tyrosine kinase inhibitor, in advanced non- small cell lung cancer harboring RET translocations Version 8/22/2018 Schema Genotyping of tumor for RET translocation (Pre-study evaluation) Register Ponatinib Progressive disease, Stable disease, partial or Unacceptable toxicity complete response Off study Continue Treatment 3 Confidential A Phase II, open-label study of ponatinib, a multi-targeted oral tyrosine kinase inhibitor, in advanced non- small cell lung cancer harboring RET translocations Version 8/22/2018 Schema ................................................................................................................................3 Table of Contents ................................................................. Error! Bookmark not defined. 1 Objectives ...................................................................................................................8 1.1 Study Design ..............................................................................................................8 1.2 Primary Objective .....................................................................................................8 -

FLT3 Inhibitors in Acute Myeloid Leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2*
Wu et al. Journal of Hematology & Oncology (2018) 11:133 https://doi.org/10.1186/s13045-018-0675-4 REVIEW Open Access FLT3 inhibitors in acute myeloid leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2* Abstract FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents. Keywords: FMS-like tyrosine kinase 3 inhibitors, Acute myeloid leukemia, Midostaurin, FLT3 Introduction RAS, MEK, and PI3K/AKT pathways [10], and ultim- Acute myeloid leukemia (AML) remains a highly resist- ately causes suppression of apoptosis and differentiation ant disease to conventional chemotherapy, with a me- of leukemic cells, including dysregulation of leukemic dian survival of only 4 months for relapsed and/or cell proliferation [11]. refractory disease [1]. Molecular profiling by PCR and Multiple FLT3 inhibitors are in clinical trials for treat- next-generation sequencing has revealed a variety of re- ing patients with FLT3/ITD-mutated AML. In this re- current gene mutations [2–4]. New agents are rapidly view, we summarized the preclinical and clinical studies emerging as targeted therapy for high-risk AML [5, 6]. on new FLT3 inhibitors, including sorafenib, lestaurtinib, In 1996, FMS-like tyrosine kinase 3/internal tandem du- sunitinib, tandutinib, quizartinib, midostaurin, gilteriti- plication (FLT3/ITD) was first recognized as a frequently nib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG mutated gene in AML [7]. -

Iclusig® (Ponatinib) 15 Mg and 45 Mg Tablets for Oral Use
CENTER FOR DRUG EVALUATION AND RESEARCH Approval Package for: Application Number: NDA 203469/S-007 & S-008 Trade Name: Iclusig® 15 mg and 45 mg tablets for oral use Generic Name: Ponatinib Sponsor: ARIAD Pharmaceuticals Approval Date: December 20, 2013 S-007 provides for revisions to the labeling. S-008 provides for the addition of a risk evaluation and mitigation strategy (REMS). CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: NDA 203469/S-007 & S-008 CONTENTS Reviews / Information Included in this NDA Review. Approval Letter X Other Action Letters Labeling X REMS X Summary Review Officer/Employee List Division Director Memo X Cross Discipline Team Leader Review Medical Review(s) X Chemistry Review(s) Environmental Assessment Pharmacology Review(s) Statistical Review(s) Microbiology Review(s) Clinical Pharmacology/Biopharmaceutics Review(s) X Other Reviews X Risk Assessment and Risk Mitigation Review(s) X Proprietary Name Review(s) Administrative/Correspondence Document(s) X CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: NDA 203469/S-007 & S-008 APPROVAL LETTER DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration Silver Spring MD 20993 NDA 203469/S-007 & S-008 SUPPLEMENT APPROVAL REMS APPROVAL ARIAD Pharmaceuticals Attention: Andrew Slugg, MS, MBA Senior Director, Regulatory Affairs 26 Landsdowne Street Cambridge, MA 02139-4234 Dear Mr. Slugg: Please refer to your Supplemental New Drug Application (sNDA) (S-007) dated November 27, 2013, received November 27, 2013, submitted under section 505(b) of the Federal Food, Drug, and Cosmetic Act (FDCA) for Iclusig® (ponatinib) 15 mg and 45 mg tablets for oral use. We acknowledge receipt of your amendments dated November 27; December 3, December 12, and December 18, 2013 (2). -

Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling While Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells
Published OnlineFirst February 14, 2013; DOI: 10.1158/1535-7163.MCT-12-0305 Molecular Cancer Cancer Therapeutics Insights Therapeutics Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling while Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells Ruwanthi N. Gunawardane, Ronald R. Nepomuceno, Allison M. Rooks, Jeremy P. Hunt, Jill M. Ricono, Barbara Belli, and Robert C. Armstrong Abstract Fms-like tyrosine kinase 3 (FLT3) is implicated in the pathogenesis of acute myeloid leukemia (AML). FLT3-activating internal tandem duplication (ITD) mutations are found in approximately 30% of patients with AML and are associated with poor outcome in this patient population. Quizartinib (AC220) has previously been shown to be a potent and selective FLT3 inhibitor. In the current study, we expand on previous observations by showing that quizartinib potently inhibits the phosphorylation of FLT3 and downstream signaling molecules independent of FLT3 genotype, yet induces loss of viability only in cells expressing constitutively activated FLT3. We further show that transient exposure to quizartinib, whether in vitro or in vivo, leads to prolonged inhibition of FLT3 signaling, induction of apoptosis, and drastic reductions in tumor volume and pharmacodynamic endpoints. In vitro experiments suggest that these prolonged effects are mediated by slow binding kinetics that provide for durable inhibition of the kinase following drug removal/clearance. Together these data suggest quizartinib, with its unique combination of selectivity and potent/sustained inhibition of FLT3, may provide a safe and effective treatment against FLT3-driven leukemia. Mol Cancer Ther; 12(4); 1–10. Ó2013 AACR. Introduction downstream signaling cascades including the Ras/ Fms-like tyrosine kinase 3 (FLT3) is a member of the MAPK, PI3K/Akt, and STAT5 pathways (1, 9–13). -
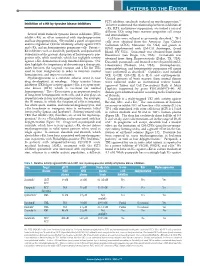
Letters to the Editor
LETTERS TO THE EDITOR FLT3 inhibitor, sorafenib, induced no myelosuppression.5,6 Inhibition of c-Kit by tyrosine kinase inhibitors To better understand the relationship between inhibition of c-Kit, FLT3, and marrow suppression, we studied a series of different TKIs using bone marrow progenitor cell assays Several small molecule tyrosine kinase inhibitors (TKIs) and immunoblots. inhibit c-Kit, an effect associated with myelosuppression Cell lines were cultured as previously described.2 TF-1 and hair depigmentation. We studied a panel of approved cells were obtained from the American Type Culture and investigational TKIs for inhibitory activity against FLT3 Collection (ATCC; Manassas, VA, USA) and grown in and c-Kit, and on hematopoietic progenitor cells. Potent c- RPMI supplemented with GM-CSF (Invitrogen, Grand Kit inhibitors such as dasatinib, pazopanib, and quizartinib Island, NY, USA). Quizartinib was obtained from Ambit demonstrated the greatest disruption of hematopoietic pro- Biosciences (San Diego, CA, USA). Crenolanib was genitor cells, while sorafenib, which has negligible activity obtained from Arog Pharmaceuticals (Dallas, TX, USA). against c-Kit, demonstrated only minimal disruption. Our Dasatinib, pazopanib, and imatinib were obtained from LC data highlight the importance of determining a therapeutic Laboratories (Woburn, MA, USA). Electrophoresis, index between the targeted receptor and c-Kit for TKIs immunoblotting, and hematopoietic progenitor cell assays used to treat malignancies in order to maintain normal were performed as described.2 Cytokines used included hematopoiesis and improve outcomes. SCF, G-CSF, GM-CSF, IL-3, IL-6, and erythropoietin. Myelosuppression is a common adverse event in new Unused portions of bone marrow from normal donors drug development in oncology. -

Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) Through Multikinase Inhibition Jessica D
Published OnlineFirst February 9, 2018; DOI: 10.1158/1078-0432.CCR-17-1928 Cancer Therapy: Preclinical Clinical Cancer Research Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) through Multikinase Inhibition Jessica D. Lang1,William P.D. Hendricks1, Krystal A. Orlando2, Hongwei Yin1, Jeffrey Kiefer1, Pilar Ramos1, Ritin Sharma3, Patrick Pirrotte3, Elizabeth A. Raupach1,3, Chris Sereduk1, Nanyun Tang1, Winnie S. Liang1, Megan Washington1, Salvatore J. Facista1, Victoria L. Zismann1, Emily M. Cousins4, Michael B. Major4, Yemin Wang5, Anthony N. Karnezis5, Aleksandar Sekulic1,6, Ralf Hass7, Barbara C. Vanderhyden8, Praveen Nair9, Bernard E. Weissman2, David G. Huntsman5,10, and Jeffrey M. Trent1 Abstract Purpose: Small cell carcinoma of the ovary, hypercalcemic type three SWI/SNF wild-type ovarian cancer cell lines. We further (SCCOHT) is a rare, aggressive ovarian cancer in young women identified ponatinib as the most effective clinically approved that is universally driven by loss of the SWI/SNF ATPase subunits RTK inhibitor. Reexpression of SMARCA4 was shown to confer SMARCA4 and SMARCA2. A great need exists for effective targeted a 1.7-fold increase in resistance to ponatinib. Subsequent therapies for SCCOHT. proteomic assessment of ponatinib target modulation in Experimental Design: To identify underlying therapeutic vul- SCCOHT cell models confirmed inhibition of nine known nerabilities in SCCOHT, we conducted high-throughput siRNA ponatinib target kinases alongside 77 noncanonical ponatinib and drug screens. Complementary proteomics approaches pro- targets in SCCOHT. Finally, ponatinib delayed tumor dou- filed kinases inhibited by ponatinib. Ponatinib was tested for bling time 4-fold in SCCOHT-1 xenografts while reducing efficacy in two patient-derived xenograft (PDX) models and one final tumor volumes in SCCOHT PDX models by 58.6% and cell-line xenograft model of SCCOHT. -

The Effects of Combination Treatments on Drug Resistance in Chronic Myeloid Leukaemia: an Evaluation of the Tyrosine Kinase Inhibitors Axitinib and Asciminib H
Lindström and Friedman BMC Cancer (2020) 20:397 https://doi.org/10.1186/s12885-020-06782-9 RESEARCH ARTICLE Open Access The effects of combination treatments on drug resistance in chronic myeloid leukaemia: an evaluation of the tyrosine kinase inhibitors axitinib and asciminib H. Jonathan G. Lindström and Ran Friedman* Abstract Background: Chronic myeloid leukaemia is in principle a treatable malignancy but drug resistance is lowering survival. Recent drug discoveries have opened up new options for drug combinations, which is a concept used in other areas for preventing drug resistance. Two of these are (I) Axitinib, which inhibits the T315I mutation of BCR-ABL1, a main source of drug resistance, and (II) Asciminib, which has been developed as an allosteric BCR-ABL1 inhibitor, targeting an entirely different binding site, and as such does not compete for binding with other drugs. These drugs offer new treatment options. Methods: We measured the proliferation of KCL-22 cells exposed to imatinib–dasatinib, imatinib–asciminib and dasatinib–asciminib combinations and calculated combination index graphs for each case. Moreover, using the median–effect equation we calculated how much axitinib can reduce the growth advantage of T315I mutant clones in combination with available drugs. In addition, we calculated how much the total drug burden could be reduced by combinations using asciminib and other drugs, and evaluated which mutations such combinations might be sensitive to. Results: Asciminib had synergistic interactions with imatinib or dasatinib in KCL-22 cells at high degrees of inhibition. Interestingly, some antagonism between asciminib and the other drugs was present at lower degrees on inhibition. -

Isoliquiritigenin, an Orally Available Natural FLT3 Inhibitor from Licorice, Exhibits Selective Anti–Acute Myeloid Leukemia Efficacy in Vitro and in Vivo
1521-0111/96/5/589–599$35.00 https://doi.org/10.1124/mol.119.116129 MOLECULAR PHARMACOLOGY Mol Pharmacol 96:589–599, November 2019 Copyright ª 2019 by The American Society for Pharmacology and Experimental Therapeutics Isoliquiritigenin, an Orally Available Natural FLT3 Inhibitor from Licorice, Exhibits Selective Anti–Acute Myeloid Leukemia Efficacy In Vitro and In Vivo Zhi-Xing Cao,1 Yi Wen,1 Jun-Lin He, Shen-Zhen Huang, Fei Gao, Chuan-Jie Guo, Qing-Qing Liu, Shu-Wen Zheng, Dao-Yin Gong, Yu-Zhi Li, Ruo-Qi Zhang, Jian-Ping Chen, and Cheng Peng Pharmacy College, Chengdu University of Traditional Chinese Medicine, Ministry of Education Key Laboratory of Standardization Downloaded from of Chinese Herbal Medicine, Key Laboratory of Systematic Research, Development and Utilization of Chinese Medicine Resources in Sichuan Province-Key Laboratory Breeding Base of Co-founded by Sichuan Province and MOST, Chengdu, China (Z.-X.C., J.-L.H., C.-J.G., S.-W.Z., D.-Y.G., Y.-Z.L., R.-Q.Z., J.-P.C., C.P.);School of Chinese Medicine, University of Hong Kong, Hong Kong, China (Y.W., F.G., Q.-Q.L., J.-P.C.); College, Shenzhen Institute of Research and Innovation, University of Hong Kong, Shenzhen, China (Y.W., F.G., Q.-Q.L., J.-P.C.); and State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, China (S.-Z.H.) molpharm.aspetjournals.org Received February 19, 2019; accepted August 20, 2019 ABSTRACT Licorice is a medicinal herb widely used to treat inflammation- AML cells. -

Characteristics of Trials and Regulatory Pathways Leading to US Approval Of
Health Policy 123 (2019) 721–727 Contents lists available at ScienceDirect Health Policy j ournal homepage: www.elsevier.com/locate/healthpol Characteristics of trials and regulatory pathways leading to US approval of innovative vs. non-innovative oncology drugs a,b,∗ b Kerstin Noëlle Vokinger , Aaron S. Kesselheim a Academic Chair for Public Law, Health Law, Digitalization and Health Policy, Faculty of Law, University of Zurich / Institute for Primary Care, University Hospital of Zurich and University of Zurich, Zurich, Switzerland b Program On Regulation, Therapeutics, and Law (PORTAL), Division of Pharmacoepidemiology and Pharmacoeconomics, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA a r a t i c l e i n f o b s t r a c t Article history: Background: Successful first-generation drugs can be converted with small alterations to second-¨ Received 17 December 2018 generation drugs,which¨ are cheaper to develop and may pose less financial risk for manufacturers due Received in revised form 22 April 2019 to already validated action mechanism and a well-defined consumer market. Accepted 4 June 2019 Methods: We found four classes of cancer drugs for first- and second generation products approved in the US: BCR-ABL tyrosine kinase inhibitors (TKI) for treatment of CML, ALK + TKI for NSCLC, CD20 monoclonal Keywords: antibodies for CLL, and HER2 monoclonal antibodies for breast cancer. We analyzed the characteristics Health policy of the clinical trials and the approval pathways for these 14 drugs. Public health Results: First-generation and 4 out of 5 s-generation BCR-ABL TKI drugs were granted expedited approval, First-generation drug while all drugs were approved based on single-arm trials. -

Precision Medicine in Pediatric Neurooncology: a Review
UCLA UCLA Previously Published Works Title Precision Medicine in Pediatric Neurooncology: A Review. Permalink https://escholarship.org/uc/item/6tv2b43d Journal ACS chemical neuroscience, 9(1) ISSN 1948-7193 Authors Mochizuki, Aaron Y Frost, Isaura M Mastrodimos, Melina B et al. Publication Date 2018 DOI 10.1021/acschemneuro.7b00388 Peer reviewed eScholarship.org Powered by the California Digital Library University of California HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author ACS Chem Manuscript Author Neurosci. Author Manuscript Author manuscript; available in PMC 2019 July 24. Published in final edited form as: ACS Chem Neurosci. 2018 January 17; 9(1): 11–28. doi:10.1021/acschemneuro.7b00388. Precision Medicine in Pediatric Neurooncology: A Review Aaron Y. Mochizuki∥, Isaura M. Frost∥, Melina B. Mastrodimos∥, Ashley S. Plant┴, Anthony C. Wang#, Theodore B. Moore∥, Robert M. Prins#,∇,○, Paul S. Weiss*,†,‡,§,∇, Steven J. Jonas*,†,∥,◆,¶ †California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California 90095, United States ‡Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California 90095, United States §Department of Materials Science and Engineering, University of California, Los Angeles, Los Angeles, California 90095, United States ∥Department of Pediatrics, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California 90095, United States ┴Division of Pediatric Oncology, Children’s Hospital of Orange -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG .